

Autor : Esquivel Florencia1, Costantini Andrea2, Maspero Jorge3, SimĂłn Gonzalo2, Moorat Anne, Mallett Steve
1Laboratorios Phoenix SAICF 2 GlaxoSmithKline Argentina 3CIDEA Argentina
https://doi.org./10.56538/ramr.ABRV4728
Correspondencia : Florencia Esquivel E-mail: floresqui07@yahoo.com.ar
ABSTRACT
Asthma is a serious worldwide health
problem. According to the last report of the MinÂistry of Health, 1,380,000
subjects suffer from asthma in Argentina. The International Guidelines (Europe,
United States, WHO [World Health Organization]) have varying approaches to
define equivalence considerations and the possibility of switching orally
inhaled products. Whereas an in vitro approach is possible, in general
the Guidelines recommend providing more clinical evidence that supports the
possibility of switching from the innovative product to another one
subsequently developed with the same acÂtive principles in the form of dry
powder inhaler. This randomized, phase IV study has been conducted to establish
the efficacy, safety and tolerability of Neumoterol®
400 compared to the reference medicinal product Symbicort
forte, budenoside/formoterol
fumarate 320/9 μg twice daily in asthmatic patients. Also, the patients’ preference on
the use of the devices has been evaluated.
The evaluated formulation has
proven to be non-inferior compared to the reference medicinal product. The
lower limit of the 95% CI (confidence interval) for the treatÂment difference
was greater than the prespecified non-inferiority
margin of –125 mL (difference: 0.044 l [95% CI: –0.008 to 0.096]). Also, higher
values were evidenced for the AUC0-10h
(area under the curve) of the FEV1 (forced
expiratory volume in the first second) and a more important change of the
baseline score in the asthma control test on day 29 for the budenoside/formoterol fumarate capsules of
400/12 μg. In one exploratory
test about the patients’ preference on the use of the devices, a higher proÂportion
of participants expressed their global preference for the budenoside/formoterol fumarate capsule of
400/12 μg. No differences
were reported in the incidence of AEs (adverse events) or SAEs (serious adverse
events) during or after the treatment. The safety profile of both products in
general coincides with the verified profile of budenoÂside/formoterol fumarate.
Key words: Asthma; Budenoside; Formoterol
fumarate; Inhalation device.
RESUMEN
El asma es un grave problema de salud mundial.
Según el último informe del Ministerio de Salud, 1 380 000
sujetos padecen asma en la Argentina. Las guías internacionales (Europa,
Estados Unidos, OMS) varían en su enfoque para definir la equivalencia y
la posibilidad de intercambio de los productos para inhalación
respiratorios. Si bien es posible un enfoque in vitro, en general las
guías recomiendan brindar más indiÂcios clínicos que avalen
la posibilidad de intercambiar el producto innovador por otro posteriormente
desarrollado con iguales principios activos en polvo seco para inhalar. Este
estudio, aleatorizado de fase IV, se realizó para establecer la
eficacia, seguridad y tolerabilidad de Neumoterol® 400 en
comparación con el producto medicinal de refeÂrencia Symbicort
forte budesonida/fumarato de formoterol 320/9 μg, indicados 2 veces al día en pacientes
asmáticos. Además, se evaluó la preferencia de los
pacientes por uno u otro dispositivo.
Se demostró la no inferioridad de la
formulación evaluada en comparación con el proÂducto medicinal de
referencia. El límite inferior del IC del 95 % para la diferencia entre los
tratamientos fue mayor que el margen predefinido de no inferioridad de –125 mL (diferencia: 0,044 l [IC del 95 %: –0,008 a 0,096]).
Asimismo, se comprobaron valores más altos para el AUC0-10h del FEV1 y un mayor cambio respecto del puntaje basal
en la prueba de control del asma el día 29 para las cápsulas de budesonida/fumarato de formoterol 400/12 μg. En un
análisis exploratorio sobre la preferencia de los pacientes por los
dispositivos, una mayor proporción de participantes expresaron su
preferencia global por la cápsula de budesonida/fumarato de formoterol 400/12 μg. No se informaÂron diferencias en la incidencia de
AE o SAE graves durante el tratamiento o después de este. El perfil de
seguridad de ambos productos en general concordó con el perfil comprobado
de budesonida/fumarato de formoterol.
Palabras clave: Asma; Budesonida; Fumarato
de formoterol; Dispositivo inhalatorio
Recibido: 06/05/2021
Aceptado: 10/03/2022
INTRODUCTION
Asthma is a chronic inflammatory disorder
of the airways, characterized by hyperreactivity of
the airway that produces recurring episodes of sibiÂlance, shortness of breath
and cough, especially at night or early in the morning. It is a high-prevalence
disease and represents an important public health problem.1, 2
The guidelines of the Global
Initiative for Asthma (GINA) highlight the need to treat the inflammation of
the airways in asthma, and also to acknowledge the importance of prophylactic
inhaled drugs, such as inhaled corticosteroids (ICS) and the combinations of
ICS/βlong-acting
beta-adrenergic agonists (LABA) (also called long-acting β-2 agonists), like the product containing the budenoside/formoterol fumarate combination (BFF).2
Symbicort forte® (with turbuhaler® inhaler) was
authorized in 2010 in Argentina for the treatment of asthma and chronic
obstructive pulmonary disease (COPD).3
Phoenix laboratory developed Neumoterol®
400, a combination of a fixed-dose of BFF dry powder (DPI), in
capsules, to be administered by means of a single-dose inhaler provided by the Plastiape Company. This formulation consists of a capsule
containing a small amount of powder with a mixÂture of 400 μg of micronized budenoside,
12 μg of micronized formoterol fumarate and
excipients. It is indicated as maintenance therapy against asthÂma and to treat
patients suffering from COPD. The International Guidelines from Europe, the
United States and the WHO, for example, have varying approaches regarding
equivalence considerations and the possibility of switching orally inhaled
products. Whereas an in vitro approach is possible, in general the
Guidelines recommend providing more clinical evidence that supports the
possibilÂity of switching between different formulations of the same active
principles. However, in areas with emerging markets, the regulatory approach of
providing commercial licensing for respiratory inhalers is generally based on
the establishment of pharmaceutical equivalence only through an in vitro analysis.
In the case of Argentina, the BFF capsule (Neumoterol® 400) was
approved only basing on in vitro evidence.
This phase IV study was conducted
to show the non-inferiority (primary objective), and gather scientific evidence
about the efficacy, safety and tolerability, and also to show the patients’
preferÂence for the BFF 400/12 μg capsule (Neumoterol®) in comparison with the reference
medicinal prodÂuct (RMP) BFF 320/9 μg (Symbicort forte®) in asthmatic patients.
METHODS
Study design
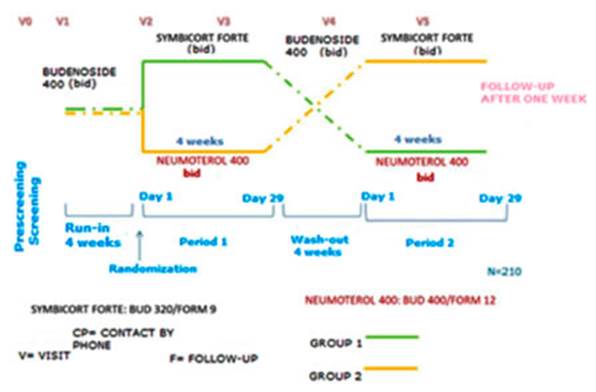
A phase IV, multicentric,
open, randomized, douÂble-crossover, non-inferiority study to compare the
efficacy, safety and tolerability of Neumoterol® 400 (BFF
400/12 μg) administered
through its specific inhaler, and the RMP, Symbicort
forte® (BFF
320/9 μg) administered
through the turbuhaler® device in adult asthmatics. (Figure 1)
The study was carried out in
accordance with the Good Clinical Practice (GCP) and the InterÂnational Council
for Harmonization (ICH) and all current requirements related to subjects’
confidenÂtiality, apart from the ethical principles detailed in the Declaration
of Helsinki of 2008. We obtained written informed consent of each subject
before specific study procedures were performed.
The study was divided in six
phases: prescreenÂing, screening/run-in (4 weeks), treatment period 1 (4
weeks), washout (at least, 4 weeks), treatment period 2 (4 weeks) and follow-up
(1 week). The total duration for each subject was at least 17 weeks. The
schedule included up to six visits and one follow-up phone call.
At the prescreening visit,
informed consent was obtained before performing any procedure or making changes
in the dosing regimen of each participant. During prescreening, patients were
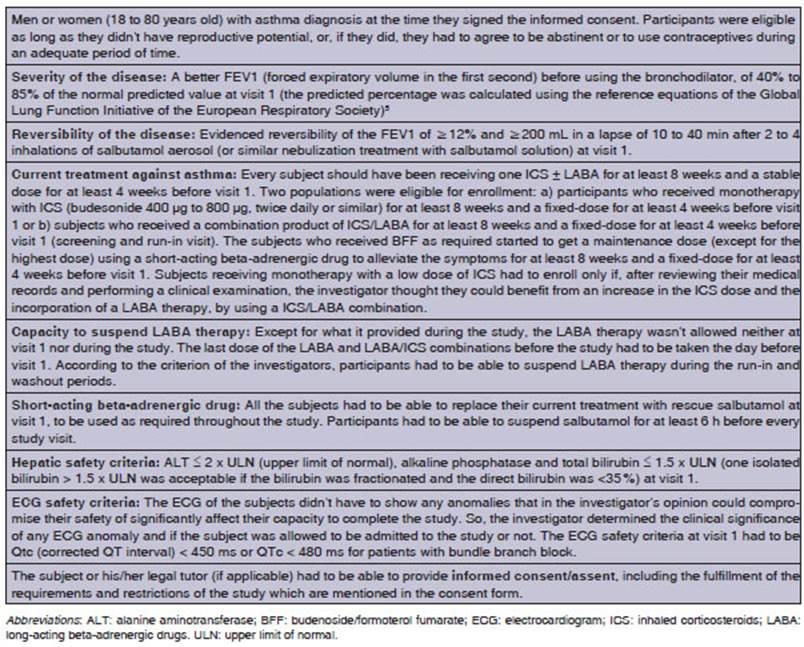
instructed about the screening and run-in visit. During visit 1 (screening and
run-in), subjects who met the inclusion criteria began a run-in period of 4
weeks. Tables 1 and 2 provide information about inclusion and exclusion
criteria.
During both the run-in and
washout periods, all the subjects received 400 μg of budenoside as DPI,
twice a day. All the participants were allowed to use rescue medication
(pressurized metered-dose inhaler of salbutamol 100 μg) during the course of the study, until visit 5.
At the end of the run-in period,
the patients were re-evaluated; each subject was told to self-administer the
study medication in each treatÂment period during 4 weeks, in the following
way, taking into account the randomization schedule:
a) inhalation of one capsule of Neumoterol® 400 (BFF
400/12 μg) through his/her
device twice daily, one capsule in the morning and the other one in the
evening/night (approximately 12 hours later); b) inhalation of RMP through turbuhaler® (BFF 320/9 μg) twice daily, in the morning and in the evening
(approximately 12 hours later). After the washout phase of 4 weeks, the
participants began the alternative treatment according to the randomÂization
schedule. The following procedures were carried out: efficacy tests, pharmacodynamic (PD) analyses, patient preference survey on
the use of the devices, and safety evaluations.
To participate in the study,
eligible subjects were required to show a better forced expiratory volume in
the first second (FEV1)
before the use of the bronchodilator between ≥ 40% and ≤ 85% of the
normal predicted value during visit 1 (screening and run-in visit). The
predicted percentage was calculated using the reference equations of the Global
Lung Function Initiative of the European Respiratory Society5
and applying equations or race adjustments, as appropriate.
Disease reversÂibility was evidenced with the improvement in the FEV1 (≥ 12%
and ≥ 200 mL) in a lapse of 10 to 40 min after 2 to 4 inhalations of
salbutamol as aerosol inhaler (or similar nebulization treatment with
salbutamol solution). Also, the FEV1
stability limit was considered as a reference point of the
subjects’ asthma status at run-in, and was used for the comparison throughout
the whole treatÂment phase to evaluate the safety of the subject. It was
calculated at visit 2 as 75% of the best FEV1 before salbutamol.
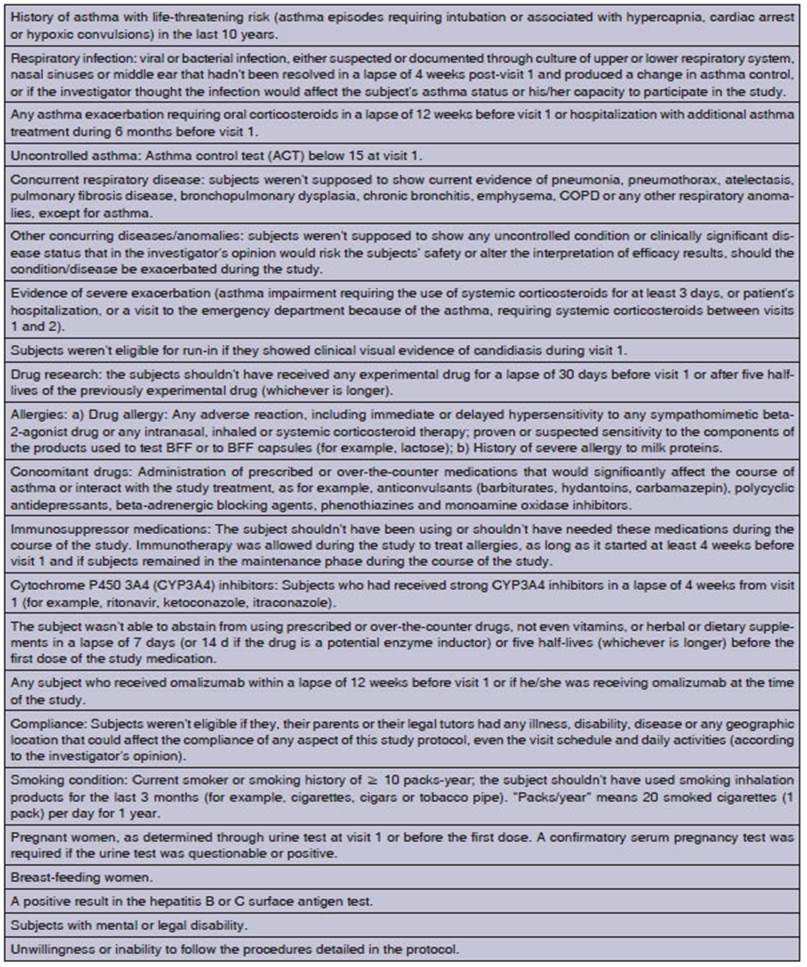
Patients were dismissed from the
study if they missed a required visit, if they didn’t show up for a re-scheduled visit or weren’t able to contact the clinic
to re-schedule the missed visit. If the particiÂpant couldn’t be contacted, the
case was considered as withdrawal by subject for a primary reason, “lost to
follow-up”. Furthermore, subjects were dismissed from the study if some of the
following criteria were met after taking into account the mean duration of the QTc of electrocardiograms in triplicate: a) QTc > 500 ms; b) QT (not
corrected) > 600 ms; c) increase of at least 60 ms compared to baseline QTc.
No privileges or
protocol exemptions were alÂlowed, except for immediate safety concerns.
Compliance: The subjects received study treatÂment at home, except
for the morning doses of visÂits 2 to 5, which they received at the clinic and
were observed by the study staff to ensure an adequate administration. Patient
compliance was evaluated during clinical visits 3 and 5 and in case of early
discontinuation by reviewing the dose counter of the
RMP device and counting unused capsules. If the compliance rate was lower than
80% or higher than 120%, the patient was re-educated about the indicated dose.
If the treatment was suspended prematurely during the course of the study or
the compliance was outside the acceptable range, the Center monitor would be
contacted for the purpose of analyzing the subject’s eligibility to continue
his/her participation in the study.
Patient compliance
was also evaluated through a phone call at the end of the second week of each
treatment period. The Center physician/staff had shown each subject the
procedure for reading the devices accurately before the beginning of the study.
Endpoints: The primary efficacy endpoint was the change in trough
FEV1 in the morning of day 29 in comparison with the baseline value. Trough
FEV1 was defined as the morning prebronchodilaÂtor
and pre-dose value, 12 hours after last evening dose (day 28), at the end of
each treatment period.
The secondary
efficacy endpoints included the area under the curve (AUC) of the FEV1 of 0-10
h at the beginning of each treatment period (0 [beÂfore the dose], 5 min, 15
min, 30 min; 1, 2, 5 and 10 h after the morning dose on day 1) and the change
in the asthma control test (ACT) score6 compared to the baseline
value after 4 weeks of each treatment period. The equipment used to obtain spirometry measurements either met the minimum performance
recommendations of the American Thoracic Society7 or exceeded them.
All the centers used their own spirometry equipment.
The highest FEV1 was recorded after three acceptable efforts. In order to
determine the AUC0-10h, the FEV1 was measured during clinical visits 2 and 4,
at 0 (before dose), 5, 10 and 20 min; and 1, 2, 5 and 10 h after the morning
dose. The ACT was an autocomplete questionnaire of 5 items which has been
developed to measure the subject’s asthma control. It could be quickly and
easily completed at the clinical practice.6
The subject’s
preference on the use of the deÂvices at the end of each treatment period was
deÂfined as an exploratory endpoint. Such preference was analyzed by means of a
questionnaire. During visit 3, the participants were asked to complete a survey
with 3 questions; during visit 5 (end of study), they
completed a survey with 4 questions.
Safety endpoints
included the change in vital signs (pulse and arterial pressure) compared to
the baseline value, the electrocardiogram and clinical biochemistry tests, the
incidence of adverse events (AEs) during each treatment period, the incidence
of asthma exacerbations (defined as worsening of asthma that requires treatment
other than the treatment of the study or rescue salbutamol; it could even
require the use of inhaled or systemic corticosteroids, a visit to the
emergency department or hospitalization), the incidence of serious asthma
exacerbations (defined as worsening of asthma that requires systemic corticosteroids
during at least 3 days, or hospitalization or a visit to the emergency
department), the incidence of oral candidiasis evaluated through tests and
early discontinuÂation.
Statistical analysis:
Sample size calculations were based on the
primary efficacy endpoint. Variability calculations were based on a previous
study8 in which the observed within-subject stanÂdard deviation was
210 mL. Assuming said value, 168 subjects would be
necessary to show the non-inferiority of the Neumoterol®
400 inhaler (BFF 400/12 μg) and RMP with BFF 320/9 μg twice
daily in asthmatic adults, taking into account a true difÂference of –50 mL
with 90% power and a one-sided significance level of 2. 5%.
The non-inferiority
margin was set at –125 mL according to the minimal clinically important difÂferences
(MCIDs) for this population of patients. It has been shown before that MCIDs for a range of asthmatic patients were 230 mL.9
In order to consider a withdrawal rate of approximately 10%, the planned number
of subjects to be randomized was 187 participants. Around 234 subjects had to
be selected and a failure rate of 20% was expected in order to reach 187
randomized subjects and to have 168 subjects completing the study.
An intention-to-treat
(ITT) analysis was used for the primary efficacy analysis. A back-up efficacy
analysis was carried out using the per-protocol population (PP). Safety
analyses were conducted with the safety population. No interim analysis was
planned for this study.
The primary efficacy
analysis was conducted with a mixed effects analysis of covariance, with the
baseline FEV1, treatment group and period as fixed effects, and the subject as
random coefficient. The non-inferiority was evaluated by examining the lower
limit of the confidence interval (one-sided significance level of 0.025) and
comparing it with the non-inferiority margin of –125 mL.
For the secondary endpoints, other comparisons were made between the product of
the study (BFF 400/12 μg) administered through a capsule inhaler and the RMP
(BFF 320/9 μg). Such comparisons were considered as backup, and no
multiplicity adjustments were applied. Current versions of the SAS software
were used.
RESULTS
Baseline data
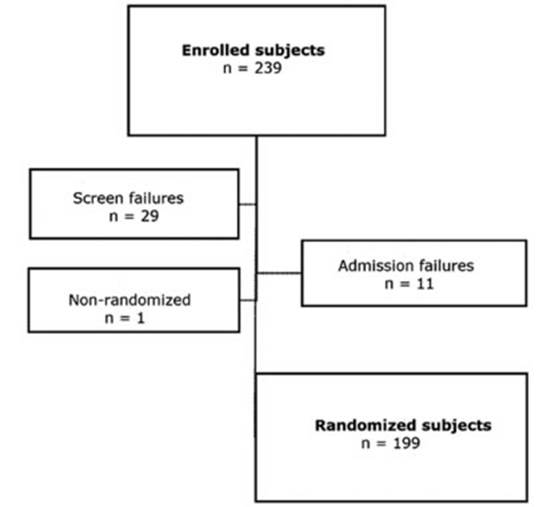
A total of 239 subjects
were enrolled in the study, and 199 were randomized (Figure 2). 184 (92%) of
the randomized subjects completed the study and 15 (8%) withdrawn from the
study. Common reasons for suspension were withdrawal of conÂsent (n = 6;
3%) and protocol deviations (n = 4; 2%). There weren’t any withdrawals
for lack of efficacy. Table 3 summarizes the distribution of the study subjects
according to each treatment period.
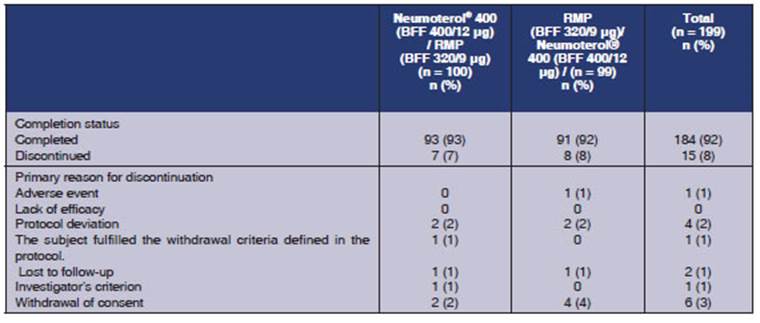
All the randomized
subjects were included in the safety and ITT population (n = 199) and 158 subÂjects
(79%) were included in the PP population.
The demographic
characteristics include both groups due to the crossover design of the study.
The participants' median of age was 47 years, and most subjects were women
(71%). Table 4 shows a complete description of baseline characteristics.
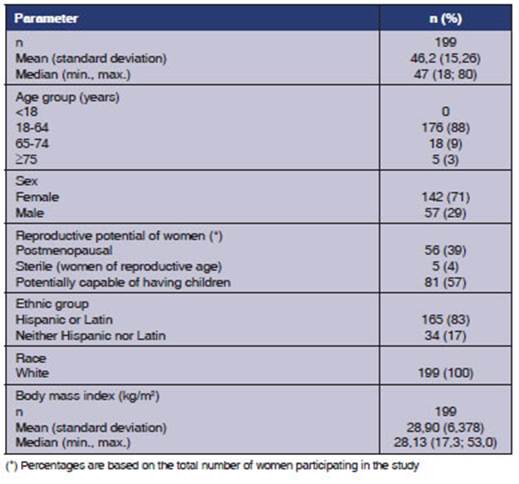
At the screening
visit, the mean FEV1 before the bronchodilator was 1.922 L (66.31% of the
normal predicted value) and the mean FEV1 after the bronchodilator was 2.393 L
(82.56% of the normal predicted value). The mean reversibility of the FEV1 was 470.60 mL (25.38%).
Cardiovascular risk
factors were reported in 49 subjects (25%), including hypertension (n =
43; 22%), hyperlipidemia (n = 10; 5%) and diabetes (n = 9; 5%).
During the study, 106 participants (53%) received one concomitant drug or more.
Analgesics, antihypertensives and antihistaminics
were the most frequently prescribed drugs.
Treatment compliance
was at least 80% for most participants (188/191 subjects [98.4%] for the Neumoterol® 400 capsule and 183/187
subjects [97.8%] for the RMP). The median of compliance was within the 94%-97%
range for both formulaÂtions during each treatment period.
Efficacy results: The primary efficacy endpoint was the change in trough
FEV1 in the morning of day
29, compared to the baseline value. In both treatments, there was an increase
in the morning trough FEV1 in
the ITT population. The mean increase in the least squares (LS) adjusted to the
model was 0.194L for Neumoterol® 400 and 0.150 L for the RMP.
The non-inferiority of the Neumoterol® 400 capsule (BFF 400/12 μg) was
demonstrated: the lower limit of the 95% confiÂdence interval (CI) for the
treatment difference was higher than the predetermined non-inferiority margin
of –125 mL (difference of 0.044 L; 95% CI: –0.008; 0.096) (Figure 3).
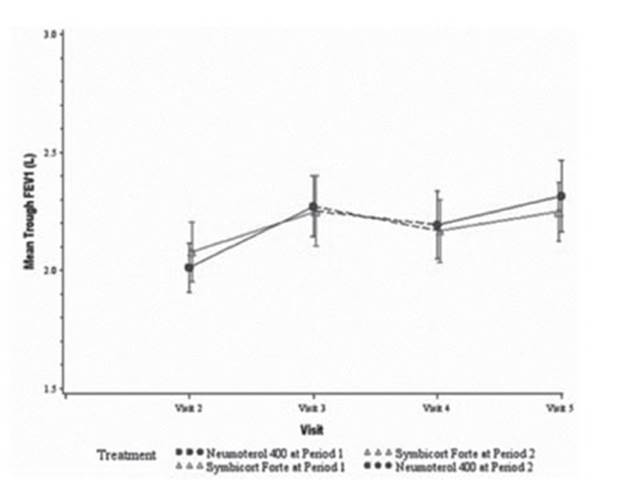
One analysis carried
out in the PP populaÂtion was similar to the primary analysis of the ITT
population. The non-inferiority of the Neumoterol®
400 capsule (BFF 400/12 μg) was evidenced when compared with the RMP (BFF 320/9
μg) (treatment difference: 0.043 l; 95% CI: –0.012;
0.098).
There was an
improvement in trough FEV1 in
both treatments from the baseline period until day 29 of period 1 (from visit 2
to visit 3) and from the baseline period until day 29 of period 2 (from visit 4
to visit 5). Whereas the trough FEV1 decreased
during the washout period of 4 weeks between peÂriods 1 and 2, it remained
above the baseline value before treatment (that is to say, the mean trough FEV1 in the baseline period for
period 2 [visit 4] was barely higher than the level observed during the
baseline period for period 1 [visit 2], regardless of the treatment).
The AUC0-10 h of the FEV1 at the beginning of each treatment
period was a secondary efficacy endpoint. In the ITT population, the AUC0-10 h of the FEV1 on day 1 was 0.98 L*h (95%
CI: 0.576; 1.384) higher for Neumoterol®
400 (BFF 400/12 μg) (Figure 4).
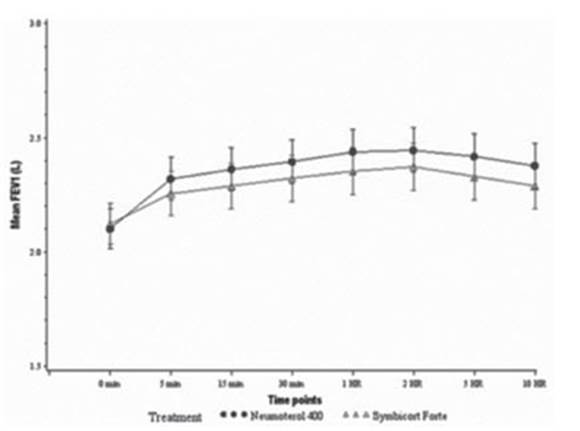
In the ITT
population, both treatments were associated with an increase in the ACT score
(another secondary efficacy endpoint) from the baseline period until day 29.
The mean increase in the LS adjusted to the model was 1.6 points for Neumoterol® 400 (BFF 400/12 μg) and
1.0 point for RMP (BFF 320/9 μg). This treatment difference favored the BFF 400/12 μg
capsule, since there was a difference of 0.6 points (95% CI: 0.1; 1.1).
Safety results: AEs were reported during treatÂment in 33 patients
(17%) of the group receiving the BFF 400/12 μg capsule
and 37 subjects (19%) of the group who received BFF 320/9 μg.
Post-treatment AEs were reported in 17 (9%) and 22 (11%) participants,
respectively. AEs (either during or after treatment) were reported in 47
subjects (24%) for the BFF 400/12 μg capsule and 51 paÂtients (26%) for BFF 320/9 μg. There
wasn’t any statistically significant difference regarding the incidence of at
least one AE neither during nor after treatment between both treatment groups
(probability index: 0.8875; 95% CI: 0.510; 1.544) (Table 5). No deaths were
reported, and none of the female participants got pregnant during the course of
the study.
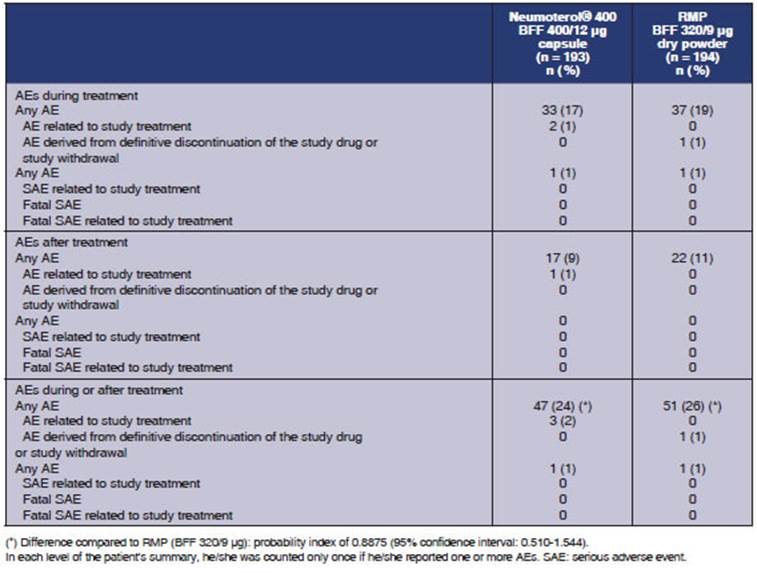
One serious AE was reported
during treatment in the BFF 400/12 μg capsule group (cholelithiasis),
and one serious AE was reported during treatment in the BFF 320/9 μg group
(rash), which led to the definitive discontinuation of the study drug. The
investigator didn’t think those serious AEs were associated with the study
drug. No post-treatment SAEs were reported. None of the subjects interÂrupted
the treatment or discontinued the study due to pre- or post-treatment AE in the
BFF 400/12 μg capsule group. The most frequently reported AEs
during treatment are summarized in Table 6. AEs reported in at least 2% of the
participants of any of the groups were: headache (4% and 3%, respectively),
rhinitis (2% and 3%) and bronchitis (1% and 3%).

The most frequently
reported post-treatment AEs were infections and infestations (4% for NeuÂmoterol® 400 [BFF 400/12 μg] and 5%
for the RMP [BFF 320/9 μg]), nervous system disorders (1% and 4%,
respectively), gastrointestinal disorders (2% and 2%), and musculoskeletal and
connective tissue disorders (2% and 0%). Headache was the only post-treatment
AE; it was reported by at least 2% of subjects (2% for BFF 400/12 μg and 0%
for BFF 320/9 μg).
The investigator
considered that two AEs durÂing treatment (palpitations and headache) and 1 AE
after treatment were associated with the BFF 400/12 μg
capsule. None of the AEs during or post-treatment were
considered to be associated with BFF 320/9 μg.
Two subjects (1%)
showed moderate asthma exacerbation during treatment with BFF 400/12 μg. One
participant (1%) showed moderate asthma exacerbation during treatment with BFF
320/9 μg. All these asthma exacerbations were resolved after
medical intervention. One patient showed moderate asthma exacerbation after
treatment, at the end of the BFF 320/9 μg treatment period, which was resolved.
No clinically
relevant differences were reÂported between the two treatments in the central
tendency for clinical biochemistry values at the baseline period or on day 29,
and no changes were reported, either, from the baseline period until day 29,
including glucose and potassium levels. No differences were observed between
the treatÂments in terms of changes in the arterial pressure or duration of the
QT interval from pre-dose until 30 min post-dose on day 29.
There was a small
increase in the heart rate, from pre-dose until 10 min post-dose on day 29 for
BFF 400/12 μg (mean increase in the LS of 1.3 beats/min), whereas
the heart rate for BFF 320/9 μg remained essentially invariable. This differÂence
between treatments regarding the change in the heart rate was statistically
significant (difference: 1.2 beats/min; 95% CI: 0.1; 2.3). The difference was
temporary, and there weren’t any differences between the BFF 400/12 μg capsule
and BFF 320/9 with respect to the change in the heart rate from pre-dose until
30 min post-dose on day 29 (difference: 0.2 beats/min; 95% CI –0.7; 1.2). No
tachycardia events were reported during the study, but one participant had an
AE of mild palpitations 3 d after the beginning of treatment with the BFF
400/12 μg capsule. No differences were observed between the
two treatments in terms of changes in systolic or diastolic arterial pressure
from pre-dose until 30 min after dose on day 29.
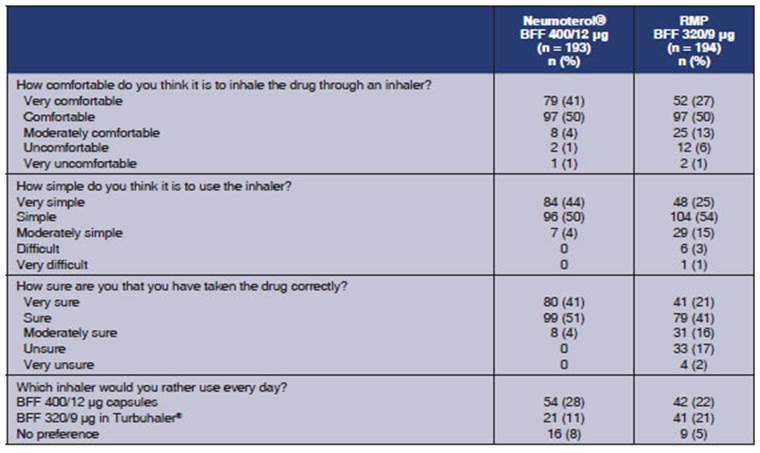
Exploratory endpoint:
The subjects’ preferÂence on the use of
the devices at the end of each treatment period was defined as an exploratory endpoint.
A higher proportion of subjects said that Neumoterol®
400 (BFF 400/12 μg) was “very comÂfortable to use” compared to the RMP
(BFF 320/9 μg), (41% versus 27%, respectively), and “very easy to
use” (44% versus 25%), and felt very confident that they had used the
medication satisfactorily (41% versus 21%). In general, more participants
expressed their preference for the Neumoterol® 400 (BFF 400/12 μg)
inhaler, 50% versus 32%. Table 7 provides a detailed description.
DISCUSSION
Asthma is a serious
worldwide health problem. There is increasing asthma prevalence in many countries,
especially in pediatric populations. This disease imposes an unacceptable
burden on healthcare systems and loss of work productivity.2
This phase IV study
demonstrated the non-inÂferiority of the efficacy of a BFF 400/12 μg capsule
inhaler (Neumoterol® 400) in comparison with the
RMP (BFF 320/9 μg) administered twice daily in asthmatic adults. BFF
400/12 μg (Neumoterol®
400) showed improvements in the parameters of the pulmonary
function and symptom control at the end of the 4-week treatment period.
It is important to
mention that no differÂence was reported between the two treatments in terms of
incidence of AEs or SAEs, neither during treatment nor after the treatment. In
general, the safety profile of both treatment strategies was similar to the one
reported before for BFF.10 Two
serious AEs were reported, but none of them was considered to be associated
with the study drug.
No difference was
reported between the two treatment strategies regarding the change from the
baseline period until day 29 in pre-dose heart rate, pre-dose arterial
pressure, predose QTc, or
the glucose or potassium levels.
It is interesting to
highlight the fact that an exploratory evaluation of the patients’ preference
on the use of the devices showed that a higher proportion of subjects expressed
global preference for the BFF 400/12 μg capsule in comparison with the inhalation of the RMP
with BFF 320/9 μg (50% versus 32%, respectively). Few studies
evaluated the preference of patients diagnosed with asthma or COPD11
with regard to inhalation devices. This aspect represents a key
factor in the improvement of treatment compliance.
This study has some
limitations: The fact that this is an open-label study could be considered a
weakness, but, to conduct a blind study with drugs administered through
inhalation where the device is not interchangeable is not possible. For that
reason, in order to improve the sensitivity of the study, a crossover design
was used instead of a parallel study. Furthermore, the open-label modality also
allowed the exploratory assessment of the patient’s preference on the type of
device.
Other limitations:
randomization per center and the performance of non-centralized spirometries with comparable yet different equipment,
accordÂing to each center.
CONCLUSIONS
The non-inferiority
of Neumoterol® 400 (BFF 400/12 μg)
evaluated in asthmatic adults was demÂonstrated, compared to the RMP (BFF 320/9
μg). A favorable tendency was observed with BFF 400/12
in the improvement of the pulmonary function on day 1 (AUC 0-10 FEV1) and in symptom control (ACT)
on day 29.
Both formulations
were well-tolerated, and their safety profile was congruent with previous
investigations. No serious AEs were reported in asÂsociation with the study
drug. A minimum (though statistically significant) change was described in the
heart rate from pre-dose until 10 min post-dose on day 29, which was bigger for
the BFF 400/12 μg capsule compared to the RMP BFF 320/9 μg; however,
this difference in the heart rate change was transitory and was no longer
observed after 30 min (difference: 0.2 beats/min; 95% CI –0.7; 1.2). A higher
proportion of patients expressed global preference for the Neumoterol®
400 capsule (BFF 400/12 μg). We believe that the study results generÂate new
clinical evidence of the safety and efficacy of this formulation, so heavily
used in Argentina.
REFERENCES
1. Mallol J, Solé D, Asher I,
et al. Prevalence of asthma symptoms in Latin America: The International Study
of Asthma and Allergies in Childhood (ISAAC). Pediatr
Pulmonol. 2000;30:439-44.
https://doi.org/10.1002/1099- 0496(200012)30:6<439::AID-PPUL1>3.0.CO;2-E
2. Global Initiative
for Asthma (GINA). Gina Report, Global Strategy for Asthma Management and
Prevention, 2016. In: www.ginasthma.org
3. Symbicort Turbohaler 400/12,
Inhalation powder.In:
https://www.medicines.org.uk/emc/medicine/11882 AcÂcessed: September 20th,
2017.
4. Argentina Census
2010 Data. In: http://www.indec.gov.ar/nivel2_default.asp?id_tema=2&seccion=P
Accessed: September 19th, 2017.
5. Quanjer PH, Stanojevic S, Cole
TJ, et al. Multi-ethnic referÂence values for spirometry
for the 3-95-yr age range: the globÂal lung function 2012 equations. Eur Respir J. 2012;40:1324- 43. https://doi.org/10.1183/09031936.00080312
6. Nathan RA, Sorkness CA, Kosinski M, et al.
DevelopÂment of the asthma control test: a survey for assessing asthma control.
J Allergy Clin Immunol.
2004;113:59-65.
https://doi.org/10.1016/j.jaci.2003.09.008
7. Miller MR,
Hankinson J, Brusasco V, et al; ATS/ERS Task Force. Standardisation
of spirometry. Eur Respir J. 2005;26:319-38.
https://doi.org/10.1183/09031936.05.00034805291
Non-Inferiority Study
of the Efficacy of Two Budenoside/Formoterol
Formulations
8. GlaxoSmithKline. GSK Clinical
Study Register. In: http:// www.gsk-clinicalstudyregister.com/study/112202#ps
AcÂcessed: Sep 21st, 2017.
9. Santanello
NC, Zhang J, Seidenberg B, et al. What are minimal important changes for asthma
measures in a clinical trial? Eur Respir
J. 1999;14:23-7 https://doi.org/10.1034/j.1399-3003.1999.14a06.x
10. Buhl R. Budesonide/formoterol for the treatment of asthma. Expert
Opin Pharmacother.
2003;4:1393-406. https://doi.org/10.1517/14656566.4.8.1393
11. Bereza
BG, Troelsgaard Nielsen A, Valgardsson
S, et al. Patient preferences in severe COPD and asthma: a comÂprehensive
literature review. Int J Chron
Obstruct Pulmon Dis. 2015;10:739-44.
https://doi.org/10.2147/COPD.S82179

| GalerĂa de imágenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|