

Autor : Maldonado Lorena, Bosio Martin, Salvado Alejandro, Chertcoff Julio
Hospital Británico de Buenos Aires
Correspondencia : Lorena Maldonado -mail: lorebcn07@hotmail.com
Resumen
A partir del 4to simposio mundial de hipertensión pulmonar (HP) se asignó un grupo propio a la hipertensión pulmonar tromboembólica crónica (HPTEC) en la clasificación de HP, el grupo 4.
La HP es un estado hemodinámico definido por una presión media de arteria pulmonar (PAPm) igual o mayor a 25 mm Hg en reposo medida por cateterismo cardiaco derecho (CCD). La HPTEC se desarrolla como consecuencia de una obstrucción en las arterias pulmonares debida a la resolución incompleta de una tromboembolia pulmonar que conduce al remodelado de los vasos pulmonares luego de tres meses de anticoagulación efectiva. En el 5to simposio mundial de HP a la HPTEC se asignó un grupo independiente de trabajo. El presente trabajo busca actualizar los datos epidemiológicos, fisiopatología, características clínicas y tratamiento de este grupo de HP.
Palabras claves: Hipertensión pulmonar; Hipertensión pulmonar tromboembólica crónica.
Abstract
Chronic thromboembolic pulmonary hypertension
Since the 4th world symposium on pulmonary hypertension (PH), chronic thromboembolic pulmonary hypertension (CTEPH) was assigned an own group in the classification of HP, the group 4.
PH is a hemodynamic state defined by a mean pulmonary artery pressure (mPAP) equal to or greater than 25 mmHg at rest, measured by right heart catheterization (CCD). CTEPH is developped by the presence of an obstruction in the pulmonary arteries due to incomplete resolution of pulmonary thromboembolic event leading to pulmonary vascular remodeling after three months of effective anticoagulation. Since the 5th World Symposium HP, CTEPH was assigned an independent working group. The objective of this paper is to update the epidemiological data, pathophysiology, clinical features and treatment of this group of pulmonary hypertension.
Key words: Pulmonary hypertension; Chronic thromboembolic pulmonary hypertension.
Introducción
La hipertensión pulmonar (HP) es un estado hemodinámico definido por una presión media de arteria pulmonar (PAPm) igual o superior a 25 mmHg en reposo, medida por cateterismo cardiaco derecho (CCD). La hipertensión pulmonar tromboembólica crónica (HPTEC) se desarrolla como consecuencia de una obstrucción en las arterias pulmonares debida a la resolución incompleta de una embolia pulmonar que conduce al remodelado de los vasos pulmonares luego de tres meses de anticoagulación efectiva1, 2.
En el 5° Congreso Mundial de HP realizado en Niza en 2013 se propusieron algunas modificaciones a la clasificación clínica que divide esta condición en 5 categorías: hipertensión arterial pulmonar (HAP) (grupo 1), HP debida a enfermedad cardiaca izquierda (grupo 2), HP debida a enfermedades respiratorias/ hipoxia (grupo 3), HP tromboembólica crónica (HPTEC) (grupo 4) e HP debida a mecanismos multifactoriales o no definidos (grupo 5)3 (Tabla 1).

La HPTEC es una enfermedad grave, potencialmente curable, donde la presencia de disnea de esfuerzo progresiva y signos de disfunción ventricular derecha pueden ser confundidos con otras patologías cardíacas o respiratorias retrasando el diagnóstico y el tratamiento4.
La HPTEC se desarrolla a consecuencia de la obstrucción del circuito vascular pulmonar debida
a uno o múltiples episodios de tromboembolismo pulmonar, con una incompleta resolución de las masas embolicas agudas, que en su evolución experimentan fibrosis y remodelación de los vasos pulmonares5. Muchos casos se producen tras fenómenos silentes de embolia de pulmón o de trombosis venosa in situ.
En las áreas que no están ocluidas por trombos, se puede desarrollar una arteriopatia similar a la de la hipertensión arterial pulmonar (HAP) y este trastorno vascular doble contribuye a la elevación de las resistencias vasculares pulmonares (RVP).
Aunque la prevalencia exacta y la incidencia anual de HPTEC son desconocidas, algunos datos sugieren que esta condición puede ocurrir en aproximadamente 5 personas por millón de habitantes por año.
En el diagnóstico diferencial de la HPTEC las siguientes patologías tienen que ser consideradas: sarcoma de arteria pulmonar, embolia tumoral, parásitos (quiste hidatídico), embolia de cuerpo extraño/congénito y estenosis adquirida de arteria pulmonar1.
Sin tratamiento, la HPTEC tiene mal pronóstico; en la actualidad puede tratarse de manera efectiva mediante cirugía, intervención conocida como endarterectomia pulmonar (EP)6. El tratamiento médico consta de anticoagulación, diuréticos y oxigenoterapia crónica en caso de insuficiencia cardíaca o hipoxemia.
Actualmente disponemos para el tratamiento médico un medicamento aprobado para HPTEC. La evidencia se ha centrado específicamente en un subgrupo de pacientes con HPTEC, aquellos con compromiso vascular pulmonar distal que los especialistas consideran inoperables o aquellos pacientes con HP o residual después de la EP3. Una técnica emergente es la angioplastia pulmonar con balón.
Incidencia
La HPTEC es evolución natural de la historia de la enfermedad tromboembólica venosa ya sea de origen agudo, subagudo o crónico. Se estima que ocurre en 0,1% al 0,5% de los que sobreviven al episodio agudo; los últimos datos publicados sugieren una incidencia un poco más elevada7-10.
Pengo y col8 describen una incidencia acumulada de HPTEC sintomática del 1% a los seis meses, 3.1% al año y del 3.8% a los dos años del seguimiento, superando la publicada por otros autores, presentándose todos los casos de HPTEC dentro de los dos años del seguimiento y el 80% de ellos en el primer año.
Korkmaz y col, en una serie de 325 pacientes, encontraron trombos residuales en el 48% de los casos a los 3 meses del tromboembolismo pulmonar (TEP), a los 6 meses del 27,4% y al año 18,2% y una incidencia de HPTEC del 4,6%11. En esta serie el 80% de los casos fueron diagnosticados dentro del primer año del evento agudo y ningún caso después del segundo año; ésto no concuerda con la hipótesis que la HPTEC se desarrolla largo tiempo después del TEP agudo, de tal modo que los autores sugieren que los casos que se presentan más tardíamente son causados por nuevas embolias que cursaron en forma asintomática. Se admite que la resolución del trombo y la restauración de la circulación pulmonar después de un TEP se producen en un tiempo de entre 4 y 8 semanas. Sin embargo, esto es reportado solo en el 50% al 80% de los casos11.
En una serie de 254 pacientes, Sánchez y col12 reportaron defectos de perfusión residual en un 29% de los casos mediante centellograma de ventilación perfusión a un año del diagnóstico de TEP.
Remy-Jardin y col13 siguieron mediante tomografía por 11 meses a 62 pacientes con TEP de arterias centrales y encontraron resolución completa en el 48%, anormalidades endovasculares en 52% e incompleta resolución en 39%; por ecocardiograma la hipertensión pulmonar persistía en solo el 8% de los casos.
Marti y col9 en una serie de 110 pacientes seguidos a 24 meses del episodio de TEP encontraron mediante ecocardiograma y arteriografía una incidencia de HPTEC del 9,1%.
En resumen, la verdadera incidencia continúa en debate, debido a que la enfermedad frecuentemente es subdiagnosticada en la práctica clínica en el contexto de una sintomatología inespecífica y una evolución variable aun en países con redes nacionales establecidas de HP4-6.
Patogenia y fisiopatología
El fallo hemodinámico y la muerte ocurre en 20% al 40% dentro de la primera hora en los pacientes con TEP agudo; en los sobrevivientes la evolución natural es la reabsorción del coágulo por acción del sistema fibrinolítico con la completa restauración del flujo6.
En un bajo porcentaje de casos, por razones no del todo conocidas, no se produce la reabsorción y la embolia evoluciona a un coágulo organizado dentro de la arteria pulmonar; esto puede ser debido a anormalidades de la hemostasia o fibrinólisis y a recurrentes embolias.
Por el momento no se ha podido confirmar una asociación significativa entre trombofilia hereditaria e HPTEC. La variación más relevante a nivel clínico es el Factor V de Leiden; se describe una fuerte relación con el riesgo de trombosis venosa profunda (TVP) y una menor asociación con la HPTEC.
Los anticuerpos antifosfolípidos están presentes entre 10-20% de los casos de HPTEC, según lo reportado por una serie de estudios4.
Las anomalías del fibrinógeno resultan interesantes en la patobiología de la HPTEC; se ha reportado una asociación entre los polimorfismos del fibrinógeno y dicha enfermedad. Numerosos factores de riesgo han demostrado incrementar la
probabilidad de HPTEC: la edad, la carga embolica inicial, la etiología idiopática del embolismo pulmonar, neoplasias, la presencia de desórdenes inflamatorios crónicos, esplenectomía, catéteres endovenosos centrales crónicos, la terapia de reemplazo tiroideo y los cortocircuitos ventrículoauriculares. El mecanismo por el que estas diferentes condiciones se asocian a HPTEC no está del todo claro aún. También se han asociado como factores de riesgo la presencia de anticoagulante lúpico y un factor VIII elevado en plasma4, 14.
Bonderman y col15 encontraron un aumento del factor VIII y anticuerpos antifosfolipídicos. Otra hipótesis es que los mecanismos fibrinolíticos están superados ya sea por la edad, el tamaño del trombo o la localización; y otros proponen que existen variantes resistentes de la fibrina a la plasmina y eso hace que se perpetúen los trombos y desarrollen HPTEC con el tiempo.
Desde hace años se admite que la simple obstrucción mecánica al flujo sanguíneo pulmonar por la falta de resolución del trombo no alcanza a explicar la HPTEC. La persistente obstrucción resulta en elevación de la presión en los vasos pulmonares y esto ocasiona estrés en las paredes de los vasos proximales a la obstrucción. Este fenómeno iniciaría una serie de eventos en los que intervendría el endotelio, mediadores inflamatorios y citoquinas que ocasionarían un remodelado de la vasculatura pulmonar o enfermedad de pequeños vasos y esto, a su vez, una mayor hipertensión y progresión de la enfermedad6.
Este modelo de “dos compartimientos” vasculares pulmonares, uno proximal y otro distal a la obstrucción fue propuesto por Moser y Braunwald luego de que el primer paciente fuera sometido a una EP en la Universidad de California en San Diego en 1970 y confirmado por estudios clínicos e histológicos que revelaron cambios en la microvasculatura similares a las observadas en el grupo 1 de HAP16, 17.
Lang y col18 avalan la hipótesis que la obstrucción macrovascular y vasoconstricción llevan a hiperflujo y éste ocasiona lesiones similares a las de la hipertensión arterial pulmonar idiopática: arteriopatía periférica, lesiones plexo génicas, micro trombosis y la hipertrofia de la media a nivel distal.
Ambos componentes, la obstrucción mecánica por el trombo y el secundario remodelado de pequeños vasos contribuyen a elevar la RVP4,6. Todos estos cambios sobre el lecho arterial pulmonar se convierten en la causa de la remodelación del corazón derecho4, 6.
Estos acontecimientos fisiopatológicos explican el empeoramiento inevitable de la HP con el tiempo y la ocasional regresión tardía o incompleta de la HP después de la EP4, 6.
Presentación clínica
La media de edad de los pacientes al momento del diagnóstico es de 63 años y el compromiso es igual en ambos sexos, siendo la población pediátrica raramente afectada1.
La presentación subclínica y la casi ausencia de signos específicos, especialmente en etapas precoces de la enfermedad, demanda un alto grado de sospecha en pacientes con disnea de esfuerzo. Muy frecuentemente el diagnóstico se demora, con una media de 14 meses entre el inicio de los síntomas y su derivación a centros expertos, en los pacientes sin historia conocida de TEP agudo y con disnea atribuida a otras patologías como insuficiencia cardíaca, asma etc.
La enfermedad se debe sospechar en pacientes con HP inexplicada con o sin historia de enfermedad tromboembólica venosa. Su presentación clínica es similar a otras variantes de HP, típicamente los pacientes se presentan de dos maneras: disnea progresiva con el ejercicio físico debida al incremento del espacio muerto y/o signos de disfunción ventricular derecho incluyendo fatiga, palpitaciones, síncope, edemas sin causa desencadenante evidente o acompañando a episodios de reembolia pulmonar4, 5, 6.
Un periodo libre de síntomas o “luna de miel” suele presentarse entre el episodio agudo de TEP y la aparición de los síntomas clínicos de HP, pudiendo ser este período de unos pocos meses a no más de un par de años. Se estima que al menos la mitad del lecho vascular pulmonar necesita estar comprometido antes de que se desarrollen los síntomas. En el 80% de los casos ocurren dentro del primer año, siendo el reporte de casos luego de ese periodo poco frecuentes8.
En el terreno diagnóstico, se recomienda la evaluación para HPTEC de los pacientes con embolia previa y disnea persistente (recomendación IIa C). Sin embargo, no se aconseja el cribado de pacientes asintomáticos tras embolia aguda (recomendación III C)19. Hasta en un 74.8% de los pacientes se
encuentra antecedente de enfermedad trombo-embólica venosa aguda20.
En estos pacientes la fatiga y la disnea progresiva durante el ejercicio son los síntomas más comunes y la clínica es indistinguible de la HAP idiopática5. A menudo, los signos clínicos en la auscultación cardíaca son sutiles: incremento del componente pulmonar del segundo ruido y un murmullo sistólico de regurgitación tricúspidea. Los signos de disfunción ventricular derecha como ingurgitación yugular, edemas, ascitis y acrocianosis se presentan en etapas más avanzadas de la enfermedad4-6.
Se describe un signo clínico patognomónico de HPTEC, un soplo en la periferia de los lóbulos inferiores resultante del flujo turbulento en áreas con flujo sanguíneo parcialmente ocluido. Este signo se encuentra en aproximadamente solo el 10% de los pacientes, siendo poco sensible pero altamente especifico5.
La mortalidad por insuficiencia cardíaca derecha en pacientes con HPTEC no diagnosticada o no tratada es similar al de otras formas de HP. La sobrevida sin intervención es pobre y proporcional al grado de HP y disfunción ventricular derecha al momento del diagnóstico. Un estudio mostró una supervivencia del 30% a los 5 años cuando la PAPm era > 40 mmHg, y del 10% cuando era > 50 mm Hg21. Esta elevada mortalidad fue confirmada por series más recientes4, 22.
Métodos de diagnóstico
La estrategia diagnóstica se debe orientar a confirmar la sospecha clínica, determinar el grado de hipertensión pulmonar e identificar a los pacientes pasibles de tratamiento quirúrgico. El diagnóstico se basa en los hallazgos obtenidos después de al menos 3 meses de anticoagulación efectiva, con objeto de discriminar esta afección de la embolia pulmonar “subaguda”. Estos hallazgos son una PAPm ≥ 25 mmHg con PCP ≤ 15 mmHg y defectos de perfusión en centellograma pulmonar ventilación perfusión1.
Los pacientes con síntomas y signos clínicos de HPTEC deben ser sometidos a estudios de imágenes para confirmar la sospecha clínica. La realización de estudios invasivos como el CCD y la angiografía pulmonar, son necesarios determinar la presencia de HP, cuantificar el nivel de la misma, definir la localización anatómica de las obstrucciones y determinar el grado y distribución de las obstrucciones vasculares pulmonares.
Diferenciar la HPTEC del tromboembolismo pulmonar recurrente es difícil porque comparten factores de riesgos, síntomas y signos clínicos. Una historia clínica minuciosa podría evidenciar una progresión gradual en la HPTEC y la presencia de exacerbaciones periódicas en el tromboembolismo pulmonar recurrente.
El ecocardiograma con Doppler es sensible para la detección de la HP y la disfunción del ventrículo derecho, pero no es específico para el diagnóstico de HPTEC.
Los hallazgos más comunes incluyen la dilatación del ventrículo derecho, la hipertrofia y la hipokinesia del ventrículo derecho, dilatación de la aurícula derecha, la sobrecarga de presión del ventrículo derecho como lo sugiere la desviación del tabique interventricular hacia la izquierda y la regurgitación tricúspide. El análisis de la velocidad de regurgitación tricúspidea permite diferenciar a los pacientes sin HP, cuya velocidad de regurgitación será < 2,8 m/seg de aquellos con HP manifiesta cuya velocidad de regurgitación es > a 3,4 m/ seg. Con ello podemos tener una aproximación de la presión sistólica de la arteria pulmonar estimada (PSAPe) a través de la ecuación de Bernoulli1, 2.
En casos excepcionales el ecocardiograma permite la visualización de los trombos en ramas proximales de la arteria pulmonar23.
Estudios recientes sugieren que la PSAPe por ecocardiografía Doppler no es tan precisa como se creía, cuando se la compara con la obtenida invasivamente mediante el CCD. Rich JD y col, mostraron una débil correlación entre ambos métodos tanto para el diagnóstico como para la evaluación de la respuesta al tratamiento24. Dentro de las recomendaciones del consenso de Niza 2013, se destaca que el centellograma pulmonar ventilación perfusión (V/Q), y no la angiotomografia pulmonar (angioTAC), es el mejor método de cribado por elección en la HPTEC.
En la figura 1 se muestra un algoritmo para el diagnóstico de HPTEC1.
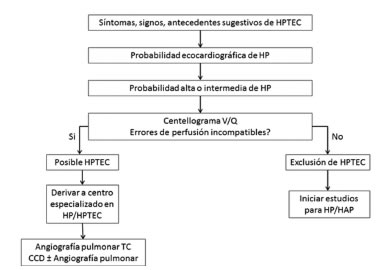
El centellograma pulmonar V/Q es la imagen de primera línea para HPTEC, ya que tiene una sensibilidad del 96-97% y una especificidad del 90-95% para el diagnóstico25. El valor predictivo negativo del V/Q es alto de tal modo que un estudio normal prácticamente descarta el tromboembolismo pulmonar crónico como causa de HP, mientras que los múltiples defectos de perfusión bilaterales sugieren fuertemente la presencia de tromboembolismo pulmonar crónico. El centellograma pulmonar V/Q no permite localizar anatómicamente la enfermedad ni su extensión y no puede ser utilizado para determinar la accesibilidad quirúrgica.
La angioTAC, tiene mucho menor sensibilidad (51%) que el V/Q para enfermedad tromboembólica crónica25, requiere de mayor experiencia en su interpretación, puede dar falsos positivos (tumores vasculares), y no detecta fácilmente enfermedad tromboembólica distal; por sí sola no permite descartar la enfermedad.
La angioTAC puede mostrar trombos excéntricos en las arterias pulmonares, densidades subpleurales, agrandamiento del ventrículo derecho, y un patrón en mosaico del parénquima si bien este hallazgo es frecuente en HPTEC, también puede observarse hasta en un 12% de pacientes con HAP26. Así mismo la angioTAC aporta datos sobre el estado del parénquima pulmonar y permite realizar diagnósticos alternativos o concomitantes y complementa la información del V/Q proporcionando datos adicionales con respecto a la localización anatómica y la accesibilidad quirúrgica23.
Suele ocurrir en la práctica clínica que los pacientes con angioTAC normal e HP suelen ser diagnosticados como de causa idiopática. En este sentido, McLaughlin y col27 describen que un 43% de los pacientes nunca se les solicitó un centellograma V/Q porque la angioTAC fue negativa. Es de importancia remarcar que se debe solicitar centellograma V/Q en todos los pacientes en estudio por HP y sospecha de origen idiopático.
La resonancia magnética nuclear tiene una sensibilidad limitada (78%) en el TEP agudo y no ha demostrado su utilidad en el diagnóstico de la HPTEC; sin embargo, sí tiene utilidad en el diagnóstico diferencial con los tumores de la arteria pulmonar que captan el gadolinio utilizado como contraste y permite diferenciarlos de los trombos.
Una herramienta esencial del diagnóstico es el CCD y la angiografía pulmonar para confirmar el diagnóstico de HP y definir la accesibilidad quirúrgica. Los valores de PAPm, la presión de oclusión arterial pulmonar y las RVP son parámetros hemodinámicos clave. En los candidatos quirúrgicos los valores pre y postoperatorios de RVP son de valor pronostico28.
Auger WR y col29, describieron en 1992 los hallazgos angiográficos característicos del TEP crónico y que aún continúan vigentes e incluyen: defectos de perfusión pulmonar de grado variable, la presencia de bandas o estenosis vasculares en donde la dilatación postestenótica es uno de los hallazgos más característicos, terminaciones vasculares abruptas en forma de amputaciones y adelgazamiento rápido o en fondo de saco. Es importante señalar que los defectos de llenado intravasculares no son frecuentes.
Se debe resaltar que pacientes con HP e imágenes compatibles con embolia pulmonar en el centellograma pulmonar V/Q o en la angioTAC deben ser derivados a centros especializados para la realización del estudio hemodinámico invasivo y la evaluación de la posibilidad del tratamiento quirúrgico. Estos centros tienen mejores resultados en el tratamiento quirúrgico y pueden ofrecer a los que no sean candidatos a cirugía una terapia invasiva o médica específica.
Tratamiento quirúrgico
La EP es el estándar de tratamiento, potencialmente curativa, aunque no siempre accesible. El mayor centro con experiencia es la Universidad de California en San Diego.
Determinar qué pacientes pueden operarse es una tarea difícil teniendo en cuenta que existen factores que influyen en esta decisión; por ejemplo, realizar un adecuado diagnóstico de HPTEC, las comorbilidades, la correlación anatómica hemodinámica y la experiencia del equipo quirúrgico30. De todos los pacientes con esta forma de HP no son candidatos a la cirugía un 36,4% según el registro internacional de HPTEC20.
Las indicaciones para la cirugía de EP incluyen: pacientes en clase funcional OMS II-IV preoperatoria, accesibilidad quirúrgica al trombo en las arterias principales, lobares y segmentarias. La edad avanzada no contraindica la cirugía ni los valores de RVP ni la presencia de disfunción ventricular derecha1. La técnica operatoria se realiza mediante una esternotomía media y bypass cardiopulmonar hipotérmico a 20°C, el objetivo fundamental es realizar una EP completa bilateral.
Esta intervención quirúrgica cuando es exitosa, al retirar los trombos que ocluyen el árbol vascular pulmonar, logra una mejoría hemodinámica inmediata, con una reducción de la PAPm de aproximadamente 50% y de la RVP de un tercio del preoperatorio.
Las complicaciones más serias de la cirugía son dos: la disfunción ventricular derecha y la lesión por isquemia de reperfusión que puede ocurrir hasta en un 15% de los pacientes1.
La mortalidad de la cirugía ha disminuido en los últimos años, se estima que en centros especializados en Europa es del 4.7% y en Estados Unidos es < 2,2% en las más recientes cirugias1, 31.
En Argentina, el Hospital Universitario Fundación Favaloro, publicó en 2011 su experiencia como único centro de referencia durante 18 años de seguimiento, con una mortalidad a 30 días y hospitalaria del 17% que ajustada a la CF esta fue diferente en las clases II-III 4%, IV 33% (p=0.01)32.
Jamieson y col33 revisaron 1500 pacientes sometidos a EP y concluyeron que ni la PAPm, ni la RVP, ni el grado de fallo ventricular derecha es un límite para la cirugía.
Sin embargo, existe una relación lineal entre la RVP preoperatoria y la mortalidad postoperatoria4. Dartevelle y col en una serie de casos publicaron que la mortalidad fue del 4% cuando la RVP preoperatoria fue menor a 900 dinas x seg/ cm5, pero se incrementó al 10% cuando la RVP se encontraba entre 900 y 1200 dinas × seg/cm5 y al 20% cuando era mayor a 1200 dinas × seg/cm5, 6. El valor de la RVP postoperatoria ha sido identificado como el principal predictor de mortalidad; cuando esta es mayor a 500 dinas x seg/cm5 la mortalidad fue del 30,6%, y cuando es menor a 500 dinas x seg/ cm5 fue del 0,9%33. Aquellos pacientes con patología inoperable (“distal”) o ante la recidiva de la HP o persistencia de la enfermedad después de la cirugía presentan un peor pronóstico.
Tratamiento médico
El tratamiento médico óptimo consiste en diuréticos, anticoagulación y oxigenoterapia crónica en caso de insuficiencia cardíaca o hipoxemia.
Aunque no existen estudios randomizados que soporten la terapia anticoagulante prolongada resulta racional indicarla para prevenir trombosis pulmonar in situ y nuevos eventos de tromboembolismo venoso.
No hay consenso en la colocación de filtros de vena cava inferior.
El tratamiento con agentes específicos vasculares pulmonares en HPTEC puede estar justificado en pacientes no quirúrgicos técnicamente o en presencia de un riesgo quirúrgico inaceptable y en pacientes con HP persistente o recurrente luego de la EP (10 al 15%).
Recientemente se publicaron los resultados del estudio clínico CHEST 1 en el que se valoró la eficacia y seguridad del riociguat, un estimulante de la guanilatociclasa, en HPTEC el mismo fue un estudio fase 3, doble ciego en 261 pacientes, de 16 semanas de duración que mostró mejoría estadísticamente significativa en el objetivo primario (distancia total recorrida en la prueba de marcha de 6 minutos, + 39m vs – 6 m); también demostró mejorías significativas en la RVP (-226 dinas.seg.cm-5 vs + 23 dinas. seg.cm-5), la clase funcional OMS, y NT-ProBNP34. Actualmente es el único fármaco aprobado para los pacientes con HPTEC inoperable o HP residual o recurrente luego de la EP.
Los otros fármacos indicados y aprobados en el grupo 1 de la HP (HAP) y utilizados de manera “off label” en la HPTEC, no han logrado demostrar el mismo nivel de evidencia; es el caso, por ejemplo del antagonista dual de los receptores de endotelina 1, bosentan (estudio BENEFIT) el que fue evaluado en 157 pacientes inoperables o con HP persistente o recurrente luego de la EP, durante 16 semanas; si bien demostró una reducción en la RVP, mejoría del índice cardiaco, reducción de los niveles de Pro-BNP y mejoría de la disnea, falló en demostrar eficacia en uno de los componentes del objetivo primario propuesto como fue la prueba de marcha de 6 minutos35.
Los estudios clínicos abiertos y de limitado número de pacientes sometidos a tratamiento con sildenafil (inhibidor de la fosfodiesterasa) y con treprostinil subcutáneo (análogo de la prostaciclina) han demostrado mejoría en la RVP, índice cardíaco,prueba de marcha de 6 minutos y en la clase funcional (OMS)23, 30.
El uso de terapias especificas en casos con compromiso hemodinámico severo como puente a la cirugía aún no cuenta con el apoyo de la evidencia científica.
Las guías recomiendan que después de la EP los pacientes deben ser seguidos en los centros de HPTEC, con al menos una evaluación hemodinámica cada 6-12 meses después de la intervención1.
Tratamiento intervencionista
En pacientes con enfermedad no accesible a la cirugía por localizarse en pequeños vasos o no candidatos por comorbilidades, la angioplastia pulmonar con balón es una opción, aunque las indicaciones concretas no se han acordado aun4.
Consiste en una técnica similar al CCD, con anestesia local, el procedimiento se realiza por etapas.
Recientemente investigadores japoneses han refinado la técnica utilizando balones más pequeños y lograron reducir el edema por reperfusion a un 2%. La angioplastia con balón está ganando la atención mundial si bien aún no es ampliamente utilizada36-38.
En pacientes muy seleccionados la mejoría hemodinámica luego del tratamiento completo parece ser similar a la obtenida por EP. Los riesgos de la técnica incluyen la perforación y la lesión por reperfusion.
Todavía no hay resultados a largo plazo y tampoco comparando con EP. Quizás algunos pacientes podrían beneficiarse de la combinación de ambos tratamientos.
Conclusiones
La HPTEC es una patología que debe ser sospechada en pacientes con disnea progresiva, intolerancia al ejercicio y signos clínicos de disfunción ventricular derecha, especialmente si tienen factores de riesgo o antecedente de enfermedad tromboembólica.
El ecocardiograma es un buen método de cribado, el centellograma V/Q y la angiotomografia permiten confirmar la sospecha diagnóstica.
El CCD permite la adecuada evaluación hemodinámica del paciente y es fundamental para conocer el grado de hipertensión pulmonar y la decisión quirúrgica.
Al momento actual la EP es el tratamiento de elección; los avances en el tratamiento médico se deben reservar para los pacientes inoperables, o con persistencia o recurrencia de la HP luego de la cirugía.
Los pacientes con HPTEC deben ser referidos a centros especializados donde podrán recibir las mejores opciones de diagnóstico y tratamiento. El estudio invasivo, el tratamiento quirúrgico, y la terapia con drogas específicas tienen mejores resultados en centros con alta experiencia en esta patología.
Conflicto de interés: Los autores del trabajo declaran no tener conflictos de intereses relacionados con esta publicación.
1. Galie N, Humbert M, Jean-Luc Vachiery et al. Guidelines for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by Association for European Paediatric and Congenital Cardiology (AEPC), International Society of Heart and Lung Transplantation (ISHLT). Eur Resp J 2015; 46: 903-975.
2. Hoeper MM, Bogaard HJ, Condliffe R et al. Definitions and diagnosis of pulmonary Hypertension. J. Am Coll Cardiol 2013; 62: D 42-50.
3. Simonneau G, Gatzoulis M, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62: S 34-41.
4. Hoeper MM, Mayer E, Simonneau G, et al. Chronic thromboembolic pulmonary hypertension. Circulation 2006; 113: 2011-2020.
5. Jenkins D, Mayer E, Screaton N, et al. State-of-the-art chronic thromboembolic pulmonary hypertension diagnosis and management. Eur Respir Rev 2012; 21: 123, 32-39.
6. Dartevelle P, Fadel E, Mussot S, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2004; 23: 637-648.
7. Fedullo P, Kerr KM, Kim NH, et al. Chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2011; 183: 1605-1613.
8. Pengo V, Lensing AWA, Prins MH, et al. Incidence of Chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350: 2257-2264.
9. Martí D, Gómez V, Escobar C, et al. Incidence symptomatic and asymptomatic chronic thromboembolic pulmonary hypertension. Arch Bronconeumol 2010 Dec; 46 (12):628-633.
10. Tapson VF, Humbert M. Incidence and prevalence of chronic thromboembolic pulmonary hypertension from acute to chronic pulmonary embolism. Proc Am Thorac Soc 2006; 3: 564-567.
11. Korkmaz A, Ozlu T, Ozlu S, et al. Long-term in acute pulmonary thromboembolism: The incidence of chronic thromboembolic pulmonary hypertension and associated risk factors. Clin Appl Thromb Hemost 2012; 18 (3): 281-288.
12. Sanchez O, Helley D, Couchon S, et al. Perfusion defects after pulmonary embolism: risk factors and clinical significance. J Thrombo Haemost 2010; 8: 1248-1255.
13. Remy-Jardin M, Louvegny S, Remy J, et al. Acute central thromboembolic disease: posttrapeutic follow-up with spiral CT angiography. Radiology 1997; 203: 173-180.
14. Sanchez O, Helley D, Couchon S, et al. Perfusion defects after pulmonary embolism: risk factors and clinical significance. J Thrombo Haemost 2010; 8: 1248-1255.
15. Bonderman D, Turecek PL, Jakowitsch J, et al. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost 2003; 90 (3): 372-376.
16. Moser KM, Braunwald NS. Successful surgical intervention in severe chronic thromboembolic pulmonary hypertension. Chest 1973; 64: 29-35.
17. Yi ES, Kim H, Ahn H, et al. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension: a morphometric and immunohistochemical study. Am J Respir Crit Care Med 2000; 162: 1577-1586.
18. Lang IM. Chronic thromboembolic pulmonary hypertension -Not so rare after all. N Engl J Med 2004; 350: 2236-2238.
19. Kostantinides S, Torbicki A, Agnelli G, et al. Guía de práctica clínica de la ESC 2014 sobre el diagnóstico y tratamiento de la embolia pulmonar aguda. Grupo de trabajo para el diagnóstico y tratamiento de la embolia pulmonar aguda de la Sociedad Europea de Cardiología (ESC) Avalado por la European Respiratory Society (ESC). Rev. Esp. Cardiol 2015; 68 (1): 64.e1-e45.
20. Pepke-Zaba J, Delcroix M, Lang I, et al. Chronic Thromboembolic Pulmonary Hypertension (CTEPH) Results From an International Prospective Registry.Circulation 2011; 124: 1973-1981.
21. Riedel M, Stanek V, Widimsky J, et al. Long-term follow-up of patients with pulmonary thromboembolism: late prognosis and evolution of hemodynamic and respiratory data. Chest 1982; 1: 151-158.
22. Ribeiro A, Lindmaker P, Johnsson H, et al. Pulmonary embolism: one-year follow-up with echocardiography doppler and five year survival analysis. Circulation 1999; 99: 1325-1330.
23. Piazza G, Goldhaber SZ. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2011; 364: 351-360.
24. Rich JD, Shah SJ, Swamy RS, et al. Inaccuracy of Doppler Echocardiographic Estimates of Pulmonary Artery Pressures in Patients With Pulmonary Hypertension: Implications for Clinical Practice. Chest 2011; 139: 988-993.
25. Tunariu N, Gibbs SJ, Win Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med. 2007; 48: 680–4.
26. Sherrick AD, Swensen SJ, Hartman TE. Mosaic pattern of lung attenuation on CT scans: frequency among patients with pulmonary artery hypertension of different causes. AJR Am J Roentgenol. 1997;169:79–82.
27. McLaughlin VV, Langer A, Tan M, et al. Contemporany trends in the diagnosis and management of pulmonary arterial hypertension: an initiative to closet he care gap. Chest 2013; 143: 324-32.
28. Skoro-Sajer N, Marta G, Gerges C, et al. Surgical specimens, haemodynamics and long-term outcomes after pulmonary endarterectomy. Thorax 2014;69: 116–122.
29. Auger WR, Fedulo PF, Moser KM, Buchbinder M, Peterson KL. Chronic major-vessels thromboembolic pulmonary artery obstruction: appearance at angiography. Radiology 1992; Feb; 182 (2): 393-8.
30. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physician, American Thoracic Society, Inc. and the Pulmonary Hypertension Association. Circulation 2009, Apr 28; 119: 2250-94.
31. Madani MM, Auger WR, Pretorius V, et al. Pulmonary endarterectomy: recent changres in a single institution´s experience of more than 2700 patients. Ann Thorac Surg 2012; 94: 97-103.
32. Favaloro R, Peradejordi M,Gomez C, et al. Tromboendarterectomia pulmonar: tratamiento de elección para la hipertensión pulmonar tromboembólica crónica. Rev Am Med Resp 2011; 2: 74-83.
33. Jamieson SW, Kapelanski DP, Sakakibara N, et al. Pulmonary endarterectomy: experience and lessons learned in 1500 cases. Ann Thorac Surg 2003; 76: 1457-1462.
34. Ghofrani HA, D´Amini AM, Grimminger F, et al. Riociguat for the treatment of Chronic Thromboembolic Pulmonary Hypertension. N Engl J Med 2013; 369: 3.
35. Jaı¨s X, D’Armini AM, Jansa P, et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in inoperable Forms of chronic Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol 2008; 52: 2127–2134.
36. Kataoka M, Inami T, Hayashida K, et al. Percutaneous transluminal pulmonary angioplasty for the treatment of chronic thromboembolic pulmonary hypertension. Circ Cardiovasc Interv 2012; 5: 756-762.
37. Mizoguchi H, Ogawa A, Munemasa M, et al. Refined balloon pulmonary angioplasty for inoperable patients with chronic thromboembolic pulmonary hypertension. Circ Cardiovasc Interv 2012; 5: 748-755.
38. Sugimura K, Fukumoto Y, Satoh K, et al. Percutaneous transluminal pulmonary angioplasty markedly improves pulmonary hemodynamics and long-term prognosis in patients with chronic thromboembolic pulmonary hypertension. Circ J 2012; 76: 485-488.

| GalerĂa de imágenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|