

Autor : SĂvori, MartĂn1-2
1 Pulmonology University Center, Faculty of Medicine. University of Buenos Aires. 2Pulmonology and Tisiology Unit. Hospital Gral. Agudos âDr.J.M.Ramos MejĂaâ. Buenos Aires
https://doi.org/10.56538/ramr.WYDL2110
Correspondencia : MartĂn SĂvori. E-mail: sivorimartin@yahoo.com
A
little history
Asthma and Chronic Obstructive
Pulmonary Disease (COPD) are heterogeneous, obstructive airway diseases, whose
physiopathology is far from being completely understood even today.
Over 60 years have passed since
the almost simultaneÂous generation of two hypotheses regarding the genesis of
asthma and chronic bronchitis associated with COPD. Despite the time that has
elapsed, with sometimes pasÂsionate and personal conflicts, they have generally
been presented as opposing hypotheses: the assertion of one denied the other,
and vice versa.1-8
More than six decades have passed
since then, and the objective of this manuscript is to review which concepts
have been confirmed over time in the light of recent research which, in the
opinion of the author of this manuscript, have been relevant.
What did the British hypothesis
say?
In the early 1950s, COPD as such
was not yet described, and Lynne McA Reid and McLean
asÂsociated smoking with the presence of bronchorÂrhea,
chronic cough, changes in bronchial mucosal defense,
bacterial colonization, and frequent infecÂtions that led to the genesis of
chronic obstructive bronchitis. This hypothesis was known as the âBritish
hypothesisâ (Figure 1).1,2

What did the Dutch hypothesis
say?
Since asthma and COPD share some
common aspects, between 1961 and 1964, Dr. NG Dick Orie
proposed in his doctoral thesis at the University of Groningen, NethÂerlands,
that asthma, chronic bronchitis, and emphysema were phenotypic expressions of
the same disease and that they evolved from one to another as individuals aged,
influenced by different factors: âBronchitis and asthma may be found in one
patient at the same age but as a rule there is a fluent development from
bronchitis in youth to a more asthmatic picture in adults, which in turn
develops into bronchitis of elderly patientsâ (Figure 2).3,
4

As a result of the interest
generated by such a bold hypotheÂsis, an International Symposium on âChronic
Bronchitisâ was organized in 1962.5 The
relationship between exogenous factors (environment, exposure to allergens, and
tobacco smoke) and endogenous factors (atopy and
bronchial hyperreactivity) would express itself
differently in chronic bronchitis. This hypothesis has caused a significant
debate among researchers from the United Kingdom, the rest of Europe, and the
United States of North America until recent years.5-10 In
1969, Dr. Fletcher labeled it as the âDutch Hypothesisâ.
MORE THAN 60 YEARS LATER, WHAT NEW INFORMATION IS AVAILABLE ABOUT THE
BRITISH HYPOTHESIS?
When the British hypothesis was
formulated, sevÂeral facts were unknown, or werenât sufficiently known as they
are today. These include, for exÂample, the evolution of the concept of
pre-COPD, the importance of the presence of respiratory symptoms in early
stages of COPD, the characÂterization of the frequent exacerbator
phenotype, and the importance of respiratory microbiota.
All of these factors currently strengthen what was stated more than 60 years
ago by Lynne McA Reid.1
Chronic respiratory symptoms
While the fundamental work of
Fletcher et al showed that chronic bronchitis (-CB- chronic cough and chronic bronchorrhea) and chronic airflow obstruction were two
separate clinical conditions that could be associated and were not related to
an accelerated loss of the lung function, more recent studies have revisited
this concept, leading to the proposal of the âPre-COPDâ stage, which will be
discussed in the following section. Bronchorrhea is
associated with a greater decline in the forced expiratory volume in the first
second (FEV1) and a higher risk of
developing COPD in young smokÂers with CB.11-14 It is also associated with a
higher number and greater severity of exacerbations.15 Several cohort studies have
examined the presÂence of chronic respiratory symptoms and their relationship
with the progression towards COPD in individuals with preserved lung function.16-22
In the SALPADIA-1 study, it was found that indiÂviduals with bronchorrhea and nearly normal lung function (GOLD 1
[Global Initiative for Chronic Obstructive Lung Disease]) experienced a higher
decline in FEV1 and
increased use of healthcare resources over a 3-year follow-up period.16
In the Copenhagen City Heart Study, it was determined that over a
5-year period, the odds ratio (OR) for the presence of bronchorrhea
as a risk factor for COPD was 1.1 (0.9-1.4), and over a 15-year period, it was
1.2 (0.9-1.6). However, bronchorrhea was associated
with a greater decline in FEV1 and
increased morbidity (hospitalization, OR of 5.3 for men [2.9-9.6] and 5.1 for
women [2.5-10.3]).17 In the SPIROMICS cohort, it
was determined that smokers with normal lung function already exhibited
increased inflammatory cellularity in bronchial mucosa compared to controls.18
In the COPDGene cohort, an accelerated
decline in lung function was observed in smokers with normal lung function.19
In the UK Biobank cohort, 351,874
subjects were studied for 9 years, examining the relationship between airflow
obstruction and the presence of respiratory symptoms.20 Among other factors, it was
determined that the deterioraÂtion in the lung function was strongly associated
with the presence of respiratory symptoms and cardiovascular comorbidities
(adjusted OR of 2, 95% confidence interval [CI] 1.91-2.14, p<0.0001, and
1.71 [1.64-1.83], p<0.0001).20 Lung function deterioration
was associated with overall mortalÂity (hazard ratio [HR]1.61 [95% CI
1.53-1.69], p <0.0001) compared to controls.20 In a study of Sherman et al of
3,948 subjects studied for 12 years, which compared patients with and withÂout
respiratory symptoms (persistent wheezing, chronic cough, chronic
expectoration, or dyspnea) in relation to FEV1
and adjusted by exposure to tobacco and height, it was observed
that men with chronic cough and women with chronic expectoraÂtion exhibited an
accelerated FEV1 decline.21
In a Copenhagen study by Lange et al, involving 13,756 subjects
studied for 10 years, it was determined that chronic expectoration was weakly
associated with overall mortality (relative risk [RR] of 1.1 for women and 1.3
for men). However, in those with severe obstruction (FEV1
of 40%), the risk was much higher (RR of 4.2).22 Currently, the therapy
recommended by the GOLD guidelines is based on the ABE matrix classification,
considering the presence of dyspnea, the degree of impairment in the quality of
life, and the type and number of exacerbations.23 However, at present, the
presence of chronic bronchorrhea or chronic cough or
the degree of bronchial obstruction are not considered in therapeutic
decisions. FEV1 is
an independent factor for mortality and has been used as an inÂclusion
criterion for the clinical development of current long-acting bronchodilators
and inhaled corticosteroids, as well as their combinations in the last 20
years.24
Anyway, recent research shows, as an example, that patients
classified as GOLD A or B with mild or severe bronchial obstruction donât have
the same disease progression. But this is not taken into account by the current
pharmacological treatment recommendations of the GOLD guideÂlines.25-26
A recent study by Han et al in individuals with tobacco exposure
(>10 pack-years) and a CAT score >10 (respiratory symptoms) showed that
dual long-acting bronchodilator therapy does not improve the quality of life.27
Pre-COPD concept
All of the historical information
mentioned above has recently been taken up by a group of renowned international
specialists who published a document where they propose to reconsider the
controversial âStage 0â concept from the 2001 GOLD guidelines and replace it
with âPre-COPDâ for patients who do not meet the current GOLD criteria for
COPD. This is based on three domains:28-30
A. Clinical symptoms: presence of
bronchorrhea, cough, dyspnea and exacerbations.
B. Functional: patients with a
post-bronchodilaÂtor FEV1/FVC
ratio greater than 0.7 but with signs of air trapping in lung volume measurements,
reduced DLCO (carbon monoxide diffusion), or signs of small airway obstruction.
C. Imaging: presence in chest
computed tomogÂraphy of centrilobular emphysema,
thickening of the bronchial walls of the large airway, or signs of involvement
of the small airway.
Microbiota and respiratory diseases
The community
of microorganisms including bacteria, fungi, viruses, and archaea
that inhabit our body collectively constitute the âhuman miÂcrobiotaâ.
If we consider the entire genetic load of these microorganisms, it is referred
to as the âhuman microbiomeâ.31-33 The
airways arenât sterÂile, and a community of microorganisms resides there,
interacting with our body in a balanced manner in a healthy state. The lower
airways have a lower biomass of microorganisms due to fewer nutrients and local
immuno mucociliary
clearance mechanisms.31-33 Microorganisms
can enter from the oropharynx through micro-aspirations or disÂpersion through
the mucosa.31-33 As a result,
the respiratory microbiota has a direct interaction
with the gut microbiota, especially that of the upper
airway.31-33 The
interaction between both microbiotas also occurs
systemically through various metabolites of the intestinal bacteria that affect
the systemic immune system. This interaction involves not only bacteria (which
are the most studied) but also the mycobiome and
pulmonary virome.31-33 When there is
an imbalÂance in this host-microorganism interaction, it is referred to as
âdysbiosisâ.31-35 Dysbiosis can be caused by antibiotics, nutritional
disruptions, or external infections that alter the benign resident commensal
flora. There has been increasingly solid evidence every year for the past two
decades that the alteration of the microbiome plays a
role in several diseases.31-35 This applies
to airway disÂeases such as asthma, COPD, or cystic fibrosis, and to other
conditions traditionally considered unrelated to microorganisms, such as
idiopathic pulmonary fibrosis, cancer, or adult respiratory distress syndrome.31-35 In the case
of asthma, exposure to microorganisms at an early age has long-term
consequences in susceptibility.36
The first generation of studies
focused on deÂscribing the genetic sequence of 16S rRNA
to charÂacterize the microbiotas of the digestive and
respiÂratory tracts. Following in vitro and in vivo studies in
animals, controlled studies in humans began to assess the host-microorganism
interaction in diseases. Some studies of multicenter academic consortiums are
currently being developed to acÂcount for population and geographic
variability, which can influence the findings, as well as method
standardization and multi-omics data analysis, going
beyond bacteria and including viruses, fungi, and archaea.
Additionally, itâs important to consider inter-individual variability in the
course of the disease and the response to various treatÂments. We are at the
start of a new era in precision medicine where the microbiome
could contribute to the understanding of new disease pathogenesis, diagnoses,
and treatments.32 COPD is a
complex syndrome characterized by different phenotypes, all sharing the common
feature of chronic airflow obstruction. The microbiota
in COPD substantially differs from that of healthy control individuals, and
this difference is even more pronounced during exacerbations.35The dynamics of these changes are influenced by multiple factors,
including the phenotypic heterogeneity of COPD, physiopathological
changes, treatments (such as corticosteroids and antibiotics), smoking, and
exacerbations.35 ApproxÂimately
40 to 50% of exacerbations are triggered by bacteria that increase airway
inflammation and obstruction, as well as bronchorrhea.
The most frequently involved bacterial genera are StreptoÂcoccus, Pseudomonas,
Moraxella, Haemophilus, Neisseria, Achromobacter, Corynebacterium,
and atypical bacteria like Mycoplasma pneumoniae and
Chlamydia pneumoniae.35 Particularly
with regard to COPD, the bacterial load is related to a higher incidence of
exacerbations and a decline in lung function.36A specific strain can generate a specific immune response, and
the appearance of a differÂent strain increases the risk of exacerbations.37 Bafadhel et al identified fundamental differences in
immunotherapy.38 The type of
exacerbation can be predicted during the stable phase of COPD. In cases of
frequent exacerbations, the pattern tends to repeat itself. The bacterial
phenotype was found in 55% of the cases, the eosinophilic in 29%, viral in 28%, and the remainder were pauci-inflammatory. IL-6 and IL-8 levels can predict among
frequent exacerbators which are the more prone to
exacerbate.38 Additionally,
viral infecÂtions can disrupt microbiome balance,
increasing susceptibility to secondary bacterial infections and associated exacerbations.31-35 Respiratory
syncytial virus, influenza A virus, and rhinovirus infections increase the
expression of bacterial adhesion molecules (e.g., ICAM-1, PAFR, CEACAM-1) on
epithelial cells, promoting the development of H. influenzae,
S. pneumoniae, and P. aeruginosa.39-40 Respiratory
viruses also deteriorate mucociliary clearance and
damage epithelial cells, disrupting the first line of defense in the
respiratory tree mucosa and allowing the invasion of pathogenic bacteria
through it.39-40 Interestingly,
it was obÂserved in animal models that the relationships between the
gastrointestinal tract microbiome and metabolites
produced by commensal bacteria in the digestive tract protect against
respiratory virus infections, while those produced by the reÂspiratory microbiome protect against bacterial and viral infections.39-40 Fungi such as
the Aspergillus genus have been
identified as etiological factors in the exacerbations of asthma, COPD, cystic
fibrosis, and bronchiectasis.31
Respiratory infections and exacerbator
phenotype
A history of severe childhood
infections is asÂsociated with decreased lung function and the presence of
respiratory symptoms in adulthood.41 There is
evidence that infection with the human immunodeficiency virus (HIV) represents
an inÂcreased risk of developing COPD (OR 1.14, 95% CI 1.05-1.25), as well as
tuberculosis.42-43 Starting with
the work of Soler Cataluña et al which clearly
demonstrated that having 1-2 exacerbations in the previous year, or even more,
compared to not having any, presented an increased risk of mortality and
hospitalization.44 Donaldson et
al showed that the subgroup of patients who experiÂence frequent exacerbations
also experience an accelerated decline in their lung function.45Furthermore, all of this was associated with a poorer quality of
life.46 The ECLIPSE
study provided additional information about how the subgroup of patients who
experienced frequent exacerbations in the previous year were more likely to
continue exacerbating over three years of follow-up, while the opposite
occurred in those who had never exÂperienced exacerbations before.47 Since 2011,
the GOLD guidelines have considered this condition as a phenotype that allowed
rating the higher risk of morbidity and mortality and conditioning the
specialized pharmacological treatment.23
MORE THAN 60 YEARS LATER, WHAT NEW INFORMATION IS AVAILABLE ABOUT THE
DUTCH HYPOTHESIS?
Many advances in the field of
genetics in asthma and COPD, the impact of neonatal development in lung
function, exposure to biomass smoke, the presence of bronchial hyperreactivity in COPD, the eosinophilic
exacerbator phenotype in COPD (or asthma-COPD
overlap), and the eosinophil as a biomarker, have or would have implications in
current management. These advancements have strengthened what was originally
proposed as the âDutch hypothesisâ more than 60 years ago by Dick Orle.3-10
Advances in genetics in asthma and COPD
The revolution in genetic research
has been one of the most marvelous and rapid advancements in understanding the
physiopathology and etiology of many diseases, including asthma and COPD, in
the last twenty years since the complete development of the Human Genome
Project.
In 2011, Dirkje
Postma revisited the Dutch hypothesis in view of the
advances in genetics and environmental factors common to both asthma and COPD.48 Based on genetic load, various enÂvironmental factors
(allergens, irritants, tobacco, etc.) triggered different rates of fetal lung
tissue growth. After birth, the relationship between genetics and environmental
factors (epigenetics) allowed for the expression of different clinical
phenotypes (Figure 3).48 More than ten
years after the formulation of the âDutch hypothesisâ, Fletcher and Peto identified in their famous study a group of
individuals who, despite exposure to tobacco smoke, would not develop COPD
(they called them ânon-susceptibleâ), while others would (âsusceptibleâ).11 Kaneko et al
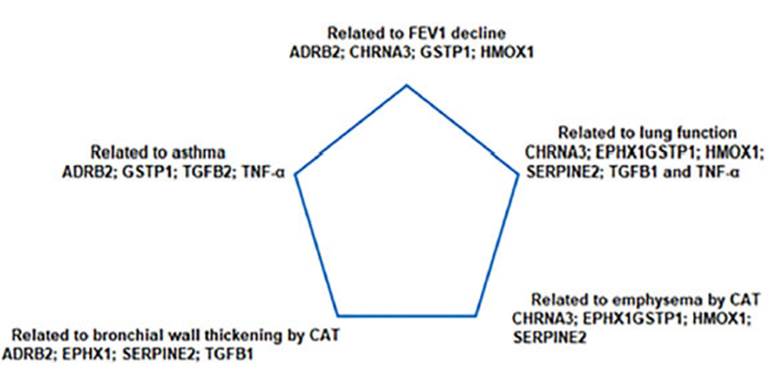
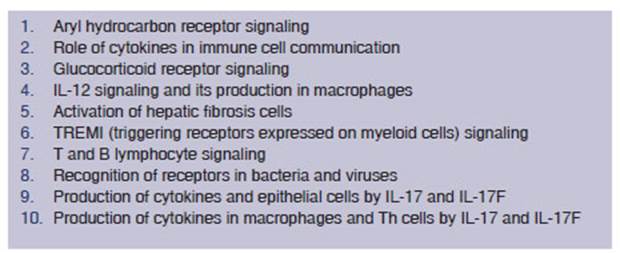
reviewed the list of coding genes common to the development of asthma and COPD.49 At least ten
molecular signaling pathways have been determined in the associated genesis of
asthma and COPD, each related to several regulator genes (Table 1).49 More
recently, Agusti and Hogg summarized the 22 genes
that are most closely associated with the development of COPD.50These are: TGFB2, PID1, RARB, EEFSEC, FAM13A, GSTCD, HHIP, TET2,
DSP, HTR4, ADAM19, AGER, ADGRG6, ARMC2, SFTPD, RIN3, THSD4, CHRNA5, CCDC101, CFÂPDP1,
MTCL1 and CYP2A6. Some of these genes are related to another famous theory that
also explains part of the physiopathology of COPD: the âproteases and antiproteasesâ theory.23, 39, 40, 50 Since the last
century, the action of proteases and the destruction of pulmonary elastic
tissue have been related to emphysema.23, 39, 40, 50 The main proteases are neutrophil elastase and proteinase-3. Serine proteases are potent
stimulators of mucus production and induce bronchorrhea
in patients with chronic bronchitis. More recently, it has been determined that
matrix metalloproteinases (MMPs) MMP-1 and MMP-9
derived from macroÂphages and neutrophils, they are overexpressed in patients
with emphysema and their synthesis is induced by the action of tobacco.23, 39, 40, 50 However, normal
lung tissue is protected against them by the activity of the antiproteases. The most significant inhibitor of serine
proteases is the alpha-1 antiÂtrypsin protein, an alpha-1 globulin. The genetic
model of emphysema caused by alpha-1 antitrypsin deficiency has been
extensively studied, and curÂrently it is possible to diagnose it early and
treat it with replacement therapy using the protein.23, 39, 40, 50 Another genetically significant factor is the
shortened length of telomeres, which is related to increased susceptibility of
the emphysema.51-52 Morla et al demonstrated in an interesting conÂtrolled
study involving normal individuals that telomere length shortens in smokers (p
= 0.05), especially those with a higher smoking load (p < 0.001), and in the
presence of bronchial obÂstruction.52 It has even
been determined which are the mutations in the telomerase-regulating gene that
have a higher risk of emphysema, idioÂpathic pulmonary fibrosis, primary bone
marrow failure, and hair loss, and these are inherited with autosomal
dominance.52
Neonatal development and lung function
The trajectory of lung function
growth is esÂtablished from gestation, birth, childhood, and adolescence.48, 53-54 50% of
patients who develop COPD may not be associated with accelerated loss of the
lung function but rather to abnormal lung growth during gestation and early
childhood.55 Genes involved
in lung development, together with maternal exposure to tobacco or biomass
smoke, have significant influence on the developÂment of asthma and COPD.48, 56-57 The
expression of different genes during the development of the uterine airway,
such as Wnt gene signaling, has been associated with
decreased lung function in childhood and asthma.58-59 The emergence of bronÂchial hyperreactivity and allergic sensitization in varying
degrees triggers inflammatory changes that contribute to structural damage of the
airway (remodeling, emphysema, small airway disease, bronchial inflammation and
bronchorrhea, etc.), ultimately leading to bronchial
obstruction and the expression of different phenotypes.50Between 4% and 12% of the general population donât have a FEV1 within the
predicted range for their gender and age. Many of them will experience airflow
limitation and accelerated loss of FEV1,
with a higher incidence at an earlier age, coexisting with heart and metabolic
diseases and higher mortality rate.60 Those who fail to reach the maximum FEV1 in early adulthood belong to a group with
higher-risk of developing COPD and other preventable and treatable diseases.61
Exposure to biomass smoke and environmental pollution
While the primary cause of COPD
in the Western world is smoking, in rural areas and urban areas without access
to natural gas, biomass combusÂtion (30-75% of which is household-based) is a
recognized factor for COPD, even in some ocÂcupational settings.23, 62-64 12% of COPD patients in the PLATINO study and
29.7% in the EPOC. AR study had no history of smoking, but 16% in PLATINO and
42% in EPOC.AR reported expoÂsure to biomass smoke.65-66 The combustion of wood, dung, charcoal, and
crop residues, releases over 250 organic compounds, volatile liquids, and
gases, where 90% are inhalable (carbon monoxÂide, ammonia, hydrocyanic acid,
formaldehyde, nitrogen and sulfur oxides, benzene, polycyclic aromatic
hydrocarbons such as benzopyrene, and kerosene).23,62-64 The risk of
COPD is 2.44 times higher in cases of exposure to biomass smoke. It has been
estimated that exposure to more than 100 hours per year is sufficient to
generate respiÂratory symptoms, and exposure to more than 200 hours per year
can lead to airflow obstruction.62-64 COPD related
to the inhalation of biomass smoke has a different phenotypic expression
compared to that of smoking. There is a greater presence of the asthma-COPD
overlap syndrome, bronchial hyperreactivity, and
bronchiectasis, less emphyÂsema, and a higher presence of chronic bronchitis
and pulmonary hypertension. In lung function tests, there is less impairment of
the FEV1, with
lower annual deterioration, and not as much imÂpairment in the DLCO test.23,62-64 As for the
relaÂtionship between the development of COPD and environmental pollution, the
American Thoracic Society has recently reviewed all the evidence and still
considers it insufficient to establish a cause-and-effect relationship.67 Regarding
asthma in children, there is strong evidence that prolonged exposure to
environmental pollution with traffic-related pollutants such as nitrogen
dioxide and black carbon is related to the onset of asthma symptoms. There is
suggestive evidence in adults as well, although it is still considered
insufficient. In asthma, environmental pollution with particles of an
aerodynamic diameter of less than 2.5 μm and ozone can lead to airway remodeling and an increase in its
incidence and severity.67
COPD and bronchial hyperreactivity
Several longitudinal studies have
demonstrated that asthma is a risk factor for the development of chronic
airflow obstruction and COPD. For example, the Tucson study showed a
twelve-fold higher risk, adjusted by smoking exposure.68Even the pattern of lung function growth in children with asthma
is associated with the development of COPD in early adulthood, a fact that had
already been anticipated by Dick Orie.3,4,69 Moreover, in the European Community
Respiratory Health Survey, bronchial hyperreactivity
(BHR) is the second independent factor mostly associated with the development
of COPD, following smoking.70 BHR is not
necessarily associated with asthma and is independently related to a higher
risk of COPD, respiratory mortality, and increased lung function decline in
mild COPD.71-72
Asthma-COPD overlap phenotype or eosinophilic exacerbator in COPD
In Spain, in 2012, Soler Cataluña et al published a document on the overlap of
asthma and COPD.73 The GESEPOC
guidelines from the same year incorporate this concept, establishing major and
minor criteria. Either two major criteria or one major criterion and two minor
criteria should be fulfilled.74 The major criteria include a highly positive bronchodilator
test (>400 ml increase in FEV1
or >15% increase), eosinophilia in sputum, and a personal
history of asthma. The minor criteÂria encompass elevated plasma levels of
total IgE (immunoglobulin E), a personal history of atopy, and a bronchodilator test showing an increase in FEV1 >200 ml
or 12% on at least two occasions.74 As from 2014,
both the GINA (Global Initiative for Asthma) and GOLD (Global Initiative for
Chronic Obstructive Lung Disease) guidelines simultaneÂously incorporated the
concept of the asthma- COPD overlap syndrome, taking into account the fact that
this subgroup of patients have a poorer quality of life, frequent
exacerbations, accelerated decline in lung function, high mortality rates, and
increased consumption of healthcare resources. Depending on the criteria that
were used, differÂent studies found a prevalence ranging from 15% to 55%.
However, in the real-world practice, it is likely to be closer to 15-20% of
patients diagnosed with these conditions. The proposed criteria inÂclude the
following: age over 40 years but with symptoms in childhood or youth;
persistent but variable exertional dyspnea; airflow
obstruction that is not completely reversible and varies over time; personal or
family history of allergies, atopy, or asthma;
symptoms that improve with treatment but may progress, requiring more
treatment; presÂence of exacerbations and comorbidities; sputum eosinophilia or
neutrophilia. The issue of diagnosÂing elderly
patients who are smokers and have a history of asthma was highlighted,
emphasizing the need for a differential diagnosis between both conditions. The
concept of asthma-COPD overlap was introduced not as a new disease but as a pheÂnotypic
expression of airway diseases, involving complex and simultaneous physiopathological mechanisms. Sin et al also published a
consensus document on the criteria for defining the asthma- COPD overlap
gathering three major criteria and at least one minor criterion: the major
criteria were persistent airflow obstruction (FEV1/FVC < 0.7 or below the lower limit of
normal) in individuals aged 40 years or older; a smoking history of at least 10
pack-years or exposure to biomass smoke or a hisÂtory of asthma before the age
of 40, or a bronchodiÂlator response of FEV1 in the spirometry
of >400 ml.76 The minor
criteria were: documented history of atopy or
allergic rhinitis; spirometry showing bronchodilator
response of FEV1 >200
ml and 12% increase compared to baseline on two or more ocÂcasions; and
eosinophilia > 300 cells/μL.76As the concept evolved, the GESEPOC guidelines stopped using the
term asthma-COPD overlap starting from the 2021 edition and explained that the exacÂerbator phenotype contains both eosinophilic
(the old asthma-COPD overlap) and non-eosinophilic
forms.77 The exacerbator phenotype was defined as any COPD patient who had
experienced two or more ambulatory exacerbations or one or more severe
exacerbations requiring hospitalization in the previous year. These
exacerbations should be separated by at least four weeks from the resoluÂtion
of the previous exacerbation or six weeks from the onset of symptoms, in order
to differentiate a new event from a relapse or therapeutic failure, considering
eosinophilia as >300 cells/μL.77
Eosinophilia as a biomarker in severe asthma and COPD + treatment
Eosinophils, as a type of cell, are involved in comÂplex roles of both innate and
adaptive immunity against infections (bacteria, viruses, fungi, and parasites)
and are also involved in the pathogenesis of neoplasms and allergic diseases.78-79 Eosinophils are multifunctional cells that interact with
various cell types (TH0 lymphocytes, basophils, endothelial cells, macrophages,
platelets, fibroblasts, and mast cells), releasing molecules and various
mediators with pro-inflammatory, cytotoxic, chemoattracÂtant,
pro-adhesive, vascular permeability-regulatÂing, and bronchoconstrictive
properties.23,
78-80 There
are different factors that can affect the variability of the eosinophil count
in peripheral blood, but it appears to have the greatest impact at higher
values and a poor impact with < 100 cells/μL.80 In
COPD, the number of eosinophils in peripheral blood
is directly related to the magnitude of the effect of inhaled corticosteroids
(ICs) in preventing exacerbaÂtions.23,
80, 82 There
wouldnât be such effect below 100 cells/μL. The greatest effect is seen above 300 cells/ μL. For counts between 100 and 300, other response predictors should be
considered.23,80,82-83 In patients
with frequent exacerbations, the GOLD guidelines suggest initiating treatment
with LAMA (long-acting muscarinic antagonist) as monotherapy
and then escalating to LAMA/LABA (long-acting beta-agonist) or LABA/ICs
combinations.23 The latter
combination is the preferred choice for patients with a history of asthma, one
severe exacerbation in the previous year, or eosinophilia >300 cells/ μL.23 Those who
have experienced more than two moderate exacerbations or one severe
exacerbation requiring hospitalization in the previous year and have eosinophil
counts greater than 100 cells/μL may be treated with
ICs/LABA.23 Patients
treated with LAMA/LABA who continue to experience exacerbations and meet these
criteria can escalate to triple therapy.23For patients with eosinophil counts between 100 and 300 cells/μL, there are factors that may predict a better response to ICs in former
smokers, experiencing exacerbations treated with systemic corticosteroids,
having more than two moderate exacerbations or one severe exacerbation, or
having coexisting asthma; on the other hand, in active smokers, a history of
pneumoÂnia or mycobacterial diseases, and exacerbations treated with
antibiotics.83
In another example of how both
hypotheses share some concepts, there is growing evidence in patients with COPD
that low eosinophil counts are associated with a higher presence of proteobacteÂria, especially Haemophilus,
and a higher incidence of bacterial infections and pneumonia.80
In severe asthma, two
inflammatory phenotypic patterns have been defined: T2-high (present in
allergic and eosinophilic asthma) and non-T2, also
called T2-low.84-85 Both T2-high
phenotypes often show some degree of overlapping. Clinical history (early
onset, family and personal history of atopic diseases), fractional exhaled
nitric oxide, increased eosinophilia, and elevated IgE
are good biomarkers of the T2-high phenotype. Allergic T2 asthma repÂresents
40-50% of severe asthma and has an atopic basis orchestrated by the activation
of T helper type 2 cells (Th2), the production of interleukins (IL) 4, IL-5,
and IL-13, and isotype switching in B lymphocytes
towards IgE production. Eosinophilic
T2 asthma represents more than 25% of severe asthma. They may be associated
with chronic rhiÂnosinusitis and nasal polyps. Severe
asthma with T2-low is characterized by low eosinophil count in peripheral blood
and sputum, with a pauciÂgranulocytic profile or neutrophilia, which may be associated with chronic airflow
limitation with air trapping and a connection to smoking.86Various monoclonal antibodies have been developed and marketed
for the T2-high phenotype.81
The first monoclonal antibody that was developed for the IgE-mediated allergic phenotype was omalizumab.
Other biologics have been developed to suppress the eosinophilic
response in patients with severe asthma (IL4, 5, and 13 inhibitors): mepolizumab and reslizumab are
IL-5 inhibitors; benralizumab is an IL-5 receptor α inhibitor, and dupilumab is an IL4 receptor α subunit inhibitor that interferes with the action of IL4 and IL13.81 In our
country, omalizumab, mepolizumab,
benralizumab, and dupilumab
are commercially available.
Unlike asthma, there arenât any
commercially available biologics yet, due to poor results in cliniÂcal studies
related to the presence of the T2-high phenotype in COPD.23, 87 Both mepolizumab (MEÂTREX and METREO studies) and benralizumab (GALATHEA and TERRANOVA studies) have not
found significant clinical benefits. There are ongoing studies with dupilumab. Various biologÂics are being tested in phase II
studies for non- T2 neutrophilic inflammation, such
as anti-IL8, etanercept and infliximab (anti-tumor
necrosis factor [TNF]-alpha), but so far, they have not achieved encouraging
results to advance to phase III studies.23, 87
CONCLUSIONS
Both hypotheses formulated over
60 years ago were initially seen as academically opposing posiÂtions. Several
decades later, in view of scientific advancements, we can affirm that they have
a strong scientific foundation that supports and complements them.39, 86 However,
there are other considerations that highlight their inaccuracies if we take
into account the current scientific knowledge. For instance, in the British
hypothesis, only a few factors (smoking and respiratory infecÂtions) were taken
into account in the genesis of chronic obstructive bronchitis. Perhaps the most
controversial aspect of the Dutch hypothesis was to consider both diseases as a
continuous evoluÂtion, despite ample evidence suggesting that, in most cases,
they are two distinct diseases, though heterogeneous with substantial clinical
and physÂiopathological differences. Itâs important to
note that there is a subgroup of patients in whom many physiopathological
and clinical aspects overlap, prompting some authors to propose for the future
a more useful, appealing, and controversial clasÂsification of chronic
obstructive diseases based on different expressed endotypes.88Far from seeing both hypotheses as antagonistic theoretical modÂels,
advancements in genetics leading to the diagÂnosis of a subtype of emphysema of
genetic origin and its replacement therapy, the understanding of the impact of
neonatal development on adult lung function, exposure to environmental biomass
and its genetic interaction, the microbiome and its
interaction with the host in relation to the physiopathology of the respiratory
disease and the exacerbations, the impact of bronchial hyperÂreactivity
and eosinophilic inflammation and their potential
impact on predicting exacerbations and the treatment of a subgroup of patients
with an exÂacerbation phenotype, as well as the metabolomics, all provide
compelling reasons to conclude that, when both hypotheses were formulated, no
one could have imagined that more than sixty years later, we would see that
both theories were right at some point and served to better understand the
genesis of asthma and COPD.39,
89
Conflict of interest
Conferences for
continuing medical education activity in asthma and COPD for Astra Zeneca, Glaxo SmithKlane and ELEA.
REFERENCES
1. Reid L. Pathology of chronic
bronchitis. Lancet. 1954;1:275-
8. https://doi.org/10.1016/S0140-6736(54)91030-2
2. MacLean K H. The histology of generalized emphysema. Australas
Ann Med. 1957;6:124-40. https://doi.org/10.1111/
imj.1957.6.2.124
3. Orie
NGM, Sluiter ID, De Vries
K, Tammeling GJ, Witkop J.
The host factor in bronchitis. In: Bronchitis. N.G.M. Orie,
H.J. Sluiter eds,
Royal Vangorcum, Assen,
1961, pp. 43-59.
4. Orie
NG. Appendix on terminology of bronchitis. In: BronÂchitis
II. N.G.M. Orie, H.J. Sluiter
eds, Royal Vangorcum, Assen, 1964, p. 398.
5. Orie
NGM, Sluiter HJ. Bronchitis: An international symÂposium.1962.
6. Postma
D, Quanjer P. In memoriam Dick Orie. Eur Respir J. 2006;891-2.
https://doi.org/10.1183/09031936.00115706
7. Postma
D, Marike Boezen H.
Rationales for the Dutch Hypothesis: Allergy and Airway Hyperresponsiveness
as genetic factors and their interaction with environÂment in the development
of asthma and COPD. Chest. 2004;126:
96S-104S. https://doi.org/10.1378/chest.126.2_suppl_1.96S
8. Hargreave
FE, Parameswaran K. Asthma, COPD and bronÂchitis are
just components of airway disease. Eur Respir J. 2006; 28: 264-7.
https://doi.org/10.1183/09031936.06.0005 6106
9. Barnes PJ. Against the Dutch
hypothesis: asthma and chronic obstructive pulmonary disease are distinct
diseases. Am J Respir Crit Care Med. 2006; 174: 240-3. https://doi.org/10.1164/rccm.2604008
10. Kraft M. Asthma and chronic
obstructive pulmonary disÂease exhibit common origins in any country. Am J Respir Crit
Care Med. 2006; 174: 238-40. https://doi.org/10.1164/rccm.2604007
11. Fletcher C, Peto R. The natural history of chronic
airflow obstruction. BMJ. 1977;1:1645-8. https://doi.org/10.1136/bmj.1.6077.1645
12. Gershon
AS, Warner L, Cascagnette P, Victor
JC, To T. Lifetime risk of developing COPD: a longitudinal populaÂtion study. Lancet. 2011;378:991-6. https://doi.org/10.1016/
S0140-6736(11)60990-2
13. Allinson
JP, Hardy R, Donaldson GC, Shaheen SO, Kuh D, Wedzicha JA. The presence of chronic mucus hypersecreÂtion
across adult life in relation to COPD development. Am J Respir Crit Care Med. 2016;193:662-72. https://doi.org/10.1164/rccm.201511-2210OC
14. Guerra S, Sherrill DL, Venker C, Ceccato CM, Halonn M, Martinez FD: Chronic
bronchitis before age 50 years preÂdicts incident airflow limitation and
mortality risk. Thorax. 2009;64:894-900.
https://doi.org/10.1136/thx.2008.110619
15. Kim V, Han MK, Vance GB, et
al. The chronic bronchiÂtis phenotype of COPD: an analysis of the COPDGene Study. Chest. 2011;140:626-33. https://doi.org/10.1378/chest.10-2948
16. Bridevaux
PO, Gerbase MW, Probst-Hensch
NM, Schindler C, Gaspoz JM, Rochat
T. Long-term decline in lung funcÂtion, utilization of care and quality of life
in modified GOLD 1 COPD. Thorax. 2008;63:768-74. https://doi.org/10.1136/thx.2007.093724
17. Vestbo
J, Prescott E, Lange P, et al. Association of chronic mucus hypersecretation
with FEV1 decline and COPD morbidity. Am J Respir Crit Care Med 1996;153:1530-5.
https://doi.org/10.1164/ajrccm.153.5.8630597
18. Woodruff PG, Barr G, Bleecker E, et al. et al Clinical signifiÂcance of symptoms
in smokers with preserved pulmonary function. New Engl
J Med. 2016;374:1811-21.
https://doi.org/10.1056/NEJMoa1505971
19. Parekh TM, Bhatia S, Cherrington A, et al. FacÂtors influencing decline in
quality of life in smokers without airflow obstruction: The COPDGene
Study. Respir Med. 2020;161:105820.
https://doi.org/10.1016/j.rmed.2019.105820
20. Higbee
DH, Dodd JW. Prevalence, risk factors and clinical implications of preserved
ratio impaired spirometry. Lancet Respir
Med. 2022;10:149-57. https://doi.org/10.1016/S2213-2600(21)00369-6
21. Sherman CB, Xu X, Speizer FE, Ferris BC,
Weiss ST, DockÂery DW. Longitudinal lung function decline in
subjects with respiratory symptoms. Am Rev Respir
Dis. 1992;146:655-9.
https://doi.org/10.1164/ajrccm/146.4.855
22. Lange P, Nyboe
J, Appleyard M, Jensen G, Schnohr
P. Relation of ventilator impairment and of chronic mucus hypersecretion
to mortality from obstructive lung disease and from all causes. Thorax. 1990;45:579-85.
https://doi.org/10.1136/thx.45.8.579
23. Global Initiative for Chronic
Obstructive Lung Disease. Global Strategy for the Diagnosis,
Management and PreÂvention of COPD 2022. Acceso el 18 Septiembre de 2022 en
https://goldcopd.org/2022-gold-reports-2/
24.
SĂvori M, FernĂĄndez R. ClasificaciĂłn de EPOC GOLD
2018: ÂżOtra oportunidad perdida? Rev Amer Med Respir. 2018;2:140-2.
25. Gedenbjerg
A, Szepligeti SK, Holm Wackerhausen
LM, et al. Prediction of mortality in patients with COPD with the new GOLD 2017
classification: a cohort study Lancet Respir Med.
2018;6:204-12. https://doi.org/10.1016/S2213-2600(18)30002-X
26.
SĂvori M, FernĂĄndez R, Toibaro
J, VelĂĄzquez Gortaire E. Supervivencia en una cohorte
de pacientes con EPOC acorde a la clasificaciĂłn GOLD 2017. Medicina Buenos
Aires. 2019:79:20-8.
27.
Han MK, Ye W, Wang D, et al. Bronchodilators in
tobacco-exposed persons with symptoms and preserved lung function. New Engl J Med. 2022;387:1173-84.
https://doi.org/10.1056/NEJMoa2204752
28. Mannino
D. GOLD Stage 0 COPD: Is it real? Does it matter? Chest.
2006;130:309-10. https://doi.org/10.1016/S0012-3692(15)51839-4
29. Vestbo
J, Lange P. Can GOLD Stage 0 provide information of prognostic value in COPD?
Am J Respir Crit Care Med.
2002;166:329-32. https://doi.org/10.1164/rccm.2112048
30.
Han M, Agusti A, Celli B,
et al. From GOLD 0 to
Pre-COPD. Am J Respir
Crit Care Med. 2021;203:414-23.
https://doi.org/10.1164/rccm.202008-3328PP
31. Rogers GB, Wesselingh S. Precision respiratory medicine and the microbiome. Lancet Respir Med.
2016;4:73-82. https://doi.org/10.1016/S2213-2600(15)00476-2
32. Rogers GB, Shaw D, Marsh R, et al. Respiratory microÂbiota:
addressing clinical questions, informing clinical practice. Thorax.
2015;70:74-81.
https://doi.org/10.1136/thoraxjnl-2014-205826
33. Anonymous editorial. Harnessing the microbiome for lung
health. Lancet Respir Med. 2019:7:827.
https://doi.org/10.1016/S2213-2600(19)30307-8
34. Budden KF, Shukla SD, Rehman SF, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019;7:1-14.
https://doi.org/10.1016/S2213-2600(18)30510-1
35. Toraldo
DM, Conte L. Influence of the lung microbiota dysbiosis in COPD exacerbations: the controversial use of
corticosteroid and antibiotic treatment and the role of eoÂsinophils
as a disease marker. J Clin Med Re. 2019;11:667- 75. https://doi.org/10.14740/jocmr3875205
36. Gloor
GB, Hummelen R, Macklaim
JM, et al. Microbiome profiling by Illumina sequencing of combinatorial sequence-tagged PCR
products. PlosOne. 2010;5:1-15.
https://doi.org/10.1371/journal.pone.0015406
37. Sethi
S, Evans N, Grant BJB, Murphy TF. New strains of bacteria and exacerbations of
COPD: New Engl J Med. 2002;347:465-71.
https://doi.org/10.1056/NEJMoa012561
38. Bafadhel
M, McKenna S, Terry S. et al. Acute exacerbaÂtions of COPD: identification of
biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184:662-71.
https://doi.org/10.1164/rccm.201104-0597OC
39. Rennard
SI. Chronic obstructive pulmonary disease: linkÂing outcomes and pathobiology
of disease modification. Proc Am Thorac
Soc 2006;3(3): 276-80.
https://doi.org/10.1513/pats.200512-129SF
40. Tam A, Din DD. Pathobiologic mechanisms of COPD. Med Clin
N Am. 2012;96:681-98.
https://doi.org/10.1016/j.mcna.2012.04.012
41.
De Marco R, Accordini S, Marcon
A, et al. Risk factors for COPD in a European cohort of young
adults: Am J Respir Crit
Care Med. 2011;183:891-7.
https://doi.org/10.1164/rccm.201007-1125OC
42. Bigna
JJ, kenne AM, Asangbeh SL, Sibetcheu AT.
Prevalence of COPD n the global population with HIV: a systematic reÂview and
meta-analysis. Lancet Glob Health. 2018;6:e193- e202. https://doi.org/10.1016/S2214-109X(17)30451-5
43. Byrne AL, Marais BJ, Mitnick CD, Lecca L, Marks GB.
Tuberculosis and chronic respiratory disease: a systemÂatic review. Int J Infect Dis. 2015;32:138-46.
https://doi.org/10.1016/j.ijid.2014.12.016
44. Soler
Cataluña JJ, MartĂnez GarcĂa
MA, RomĂĄn SĂĄnÂchez P, Salcedo
E, Navarro M, Ochando R. Severe acute exacerbations
and mortality in patients with COPD. Thorax. 2005;60:925-31. https://doi.org/10.1136/thx.2005.040527
45. Donaldson GC, Seemungal TA, Bhowmik A, et al.
RelationÂship between exacerbation frequency and lung function decline in
chronic obstructive pulmonary disease. Thorax. 2002;57:847-52. https://doi.org/10.1136/thorax.57.10.847
46. Spencer S, Calverley PMA, Burge PS: Impact of preventing exacerbations
on deterioration of health status in COPD: Eur Respir J. 2004;23:698-702.
https://doi.org/10.1183/09031936.04.00121404
47.
Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128-38.
https://doi.org/10.1056/NEJMoa0909883
48. Postma
D, Kerkhof M, Boezen M, Koppelman G. Asthma and Chronic Obstructive Pulmonary
Disease: common genes, common environments? Am J Respir
Crit Care Med. 2011;183:1588-94.
https://doi.org/10.1164/rccm.201011-1796PP
49.
Kaneko Y, Yatagai Y, Yamada H, et al. The search for comÂmon pathways underlying asthma and COPD. Int J COPD 2013;8:65-78.
https://doi.org/10.2147/COPD.S39617
50. Agusti
A, Hogg J. Update on the pathogenes of ChronÂic
Obstructive Pulmonary Disease. New Engl J Med. 2019;381:1248-56. https://doi.org/10.1056/NEJMra1900475
51. Alder J, Guo
N, Kembou F, et al. Telomere length is a determinant
of emphysema susceptibility. Am J Respir Crit Care Med. 2011;184:904-11.
https://doi.org/10.1164/rccm.201103-0520OC
52. MorlĂĄ
M, Busquets X, Pons J, Sauleda
J, MacNee W, Agusti AGN. Telomere shortening in smokers with and without COPD. Eur Respir J. 2006;27:525-8. https://doi.org/10.1183/09031936.06.00087005
53. Barker DJ, Godfrey KM, Fall
C, Osmond C, Winter Pd, Shaheen
SO. Relation of birth weight and childhood reÂspiratory
infection to adult lung function and death from chronic airways disease.
BMJ. 1991; 303:671-5.
https://doi.org/10.1136/bmj.303.6804.671
54. MartĂnez
F. Early-life origin of Chronic Obstructive PulÂmonary Disease. New Engl J Med. 2016;375;871-8.
https://doi.org/10.1056/NEJMra1603287
55. Lange P, Celli B, AgustĂ A, et al. Lung function trajectoÂries leading to
chronic obstructive pulmonary disease. N Engl J Med.
2015;373:111-22. https://doi.org/10.1056/NEJMoa1411532
56. Boy E, Bruce N, Delgado H.
Birth weight and exposure to kitchen wood smoke during pregnancy in rural
Guatemala. Environ Health Perspect. 2002;110:109-14. https://doi.org/10.1289/ehp.02110109
57. Sunyer
J, Plana E, Dharmage S, Heinrich J, Jarvis D, DeMarco R, Norback D, Raherison C, Villani S, et al. Early life origins of chronic obstructive pulmonary disÂease.
Thorax. 2010;65: 14-20.
https://doi.org/10.1136/thx.2008.112136
58. Postma
DS, de Vries K, Koeter GH, Sluiter HJ. IndepenÂdent influence of
reversibility of air-flow obstruction and nonspecific hyperreactivity
on the long-term course of lung function in chronic air-flow obstruction.
Am Rev Respir Dis. 1986;134:276-80.
59. Sharma S, Tantisira
K, Carey V, Murphy AJ, Lasky-Su J, Celedon JC, Lazarus R, Klanderman
B, Rogers A, Soto- Quiros M, et al. A role for Wnt signaling genes in the pathoÂgenesis
of impaired lung function in asthma. Am J Respir
Crit Care Med. 2010;181:328-36.
https://doi.org/10.1164/rccm.200907-1009OC
60. AgustĂ
A, Noell G, Brugada J, Faner R. Lung function in early adulthood and health in
later life: a transgeneratio cohort analysis. Lancet Respir Med. 2017; 5:935-45.
https://doi.org/10.1016/S2213-2600(17)30434-4
61. AgustĂ
A, Faner R. Lung function trajectories in health and
disease. Lancet Respir Med. 2019;7:358-64.
https://doi.org/10.1016/S2213-2600(18)30529-0
62.
Juneman A, Legarreta G.
Inhalación de humo de leña: una causa relevante pero poco reconocida de EPOC. Rev Arg Med
Respir. 2007;2:51-7.
63.
Silva R, Oyarzun M, Olloquepui J.et al. Mecanismos paÂtogĂ©nicos
en la EPOC causada por exposiciĂłn a humo de biomasa.Arch
Bronconeumol. 2015;51:285-92.
https://doi.org/10.1016/j.arbres.2014.10.005
64.
Torres Duque CA, GarcĂa RodrĂguez MC, GonzĂĄlez GarcĂa;.
EPOC por humo de leña: ¿un fenotipo deiferente o una
entidad distinta? Arch Bronconeumol.
2016;52:425-31. https://doi.org/10.1016/j.arbres.2016.04.004
65.
Menezes AM, PĂ©rez Padilla R, Jardim
JR, et al. COPD in five Latin American cities (the PLATINO
Study): a prevalence study. Lancet. 2005;365:1875-81. https://doi.org/10.1016/S0140-6736(05)67632-5
66.
Echazarreta A, Arias S, del Olmo R, et al.
Prevalencia de EPOC en 6 aglomerados urbanos de Argentina: el estudio EPOC.AR. Arch Bronconeumol. 2018;54:260-9. https://doi.org/10.1016/j.arbres.2017.09.018
67.
Thurston G. Balmes JR, Garcia E, et al. Outdoor air pollution and new onset airway disease: An Official American
Thoracic Society Workshop Report. Ann Am Thorac Soc.
2020;17:397-98.
https://doi.org/10.1513/AnnalsATS.202001-046ST
68. Silva GE, Sherrill DL, Guerra
S, Barbee RA: Asthma as a risk factor for COPD in a longitudinal study. Chest. 2004;126:59-65.
https://doi.org/10.1378/chest.126.1.59
69. McGeachie
MJ, Yates KP, Zhou X, et al. Patterns of growth and decline in lung function in
persistent childhood asthma. N Engl J Med. 2016;374:1842-52. https://doi.org/10.1056/NEJMoa1513737
70. Hospers JJ, Postma DS, Rijcken B, et al.
Histamine airway hyperresponsiveness and mortality
from chronic obstructive pulmonary disease: a cohort study. Lancet.
2000;356:1313- 7. https://doi.org/10.1016/S0140-6736(00)02815-4
71. Rijcken
B, Schouten JP, Weiss ST, Speizer FE, Van der Lende R. The relationship of nonspecific
bronchial response to respiratory symptoms in a random population sample.
Am Rev Respir Dise.
1987;136:62-8. https://doi.org/10.1164/ajrccm/136.1.62
72. Tashkin
DP, Altose MD, Connett JE,
et al. Methacholine reactivity predicts changes in
lung function over time in smokers with early chronic obstructive pulmonary
disease. The Lung Health Study Research Group. Am J Respir Crit Care Med. 1996;153:1802-11. https://doi.org/10.1164/ajrccm.153.6.8665038
73.
Soler-Cataluna JJ, CosĂo B, Izquierdo JL,
LĂłpez-Campos JL, MarĂn JM, AgĂŒero R, et al. Documento de consenso sobre el
fenotipo mixto EPOC-asma en la EPOC. Arch Bronconeumol. 2012;48:331-7.
https://doi.org/10.1016/j.arbres.2011.12.009
74.
Miravitlles M, Soler Cataluña JJ, Calle M, et al.
GuĂa espaÂñola de la EPOC (GesEPOC) Tratamiento
farmacolĂłgico de la EPOC estable. Arch Bronconeumol. 2012;48:247-57.
https://doi.org/10.1016/j.arbres.2012.04.001
75. Global Strategy for Asthma
Management and PrevenÂtion, Global Initiative for Asthma (GINA). Diagnosis of
Asthma, COPD and Asthma-COPD overlap syndrome (ACOS). 2015. Global Initiative for Asthma website. Acceso el 27 de
Septiembre de 2022 en http://www.ginÂasthma.org/.
76.
Sin JD, Miravitlles M, Mannino
DM, et al. What is asthma- COPD overlap syndrome? Towards a consensus definition from a around table discussion. Eur
Respir J. 2016;48:664- 73.
https://doi.org/10.1183/13993003.00436-2016
77.
Miravitlles M, Calle M, Molina J, et al.
ActualizaciĂłn de la GuĂa Española de la EPOC (GesEPOC).Tratamiento
farmacolĂłgico. Arch Bronconeumol. 2022;58:69-81.
https://doi.org/10.1016/j.arbres.2021.03.005
78. Asano K, Ueki S, Tamari M, et
al. Adult-onset eosinoÂphilic airway diseases. Allergy. 2020;75:3087-99.
https://doi.org/10.1111/all.14620
79. Tefferi
A. Blood eosinophilia: a new paradigm in disÂease, classification, diagnosis
and treatment. Mayo
Clin Proc. 2005;80:75-83. https://doi.org/10.1016/S0025-6196(11)62962-5
80.
Singh D, AgustĂ A, MartĂnez F, et al. Blood eosinophils and COPD: A Global Initiative for Chronic
Obstructive Lung Disease Science Committee: 2022 review. Am J Respir Crit Care Med. 2022;206:17-24. https://doi.org/10.1164/rccm.202201-0209PP
81.
SĂvori M, Pascansky D. Asma
grave T2 alto: anĂĄlisis del diseño de los estudios clĂnicos de los nuevos
biolĂłgicos. Rev Am Med Respir. 2022;1:98-115.
82. Mycroft K, Krenke R. Eosinophils in
COPD-Current concepts and clinical implications. J Allergy Clin ImÂmunol Pract.
2020;8:2565-74.
https://doi.org/10.1016/j.jaip.2020.03.017
83. Stolz
D, Miravitlles M. The right treatment for the right
paÂtient with COPD: lessons from the IMPACT trial. Eur
Respir J.
2020;55:2000881https://doi.org/10.1183/13993003.00881-2020
84. Edris
A, De Feyter S, Maes T, Joos G, Lahousse L. MonoÂclonal
antibodies in type 2 asthma: systematic review and network meta-analysis. Respiratory Research. 2019; 20: 179.
https://doi.org/10.1186/s12931-019-1138-3
85. Agache
I, Beltran J, Akdis C, et al. Efficacy and safety of
treatment with biologics (benralizumab, dupilumab, mepolizumab, omalizumab and reslizumab) for
severe eosinophilic asthma. Allergy.
2020;75:1023-42. https://doi.org/10.1111/all.14221
86. Kuo
CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, et
al; U-BIOPRED Study Group. T-helper cell type 2 (Th2) and non-Th2 molecular
phenotypes of asthma using sputum tranÂscriptomics in
U-BIOPRED. Eur Respir J.
2017;49:1602135.
https://doi.org/10.1183/13993003.02135-2016
87. Donovan T, Milan SJ, Wang R, Bancho E, Bradley P, Crossingham
I. Anti-IL-5 therapies for chronic obstrucÂtive pulmonary disease. Cochrane
Database Sys Rev. 2020;12:CD013432.
https://doi.org/10.1002/14651858.CD013432.pub2
88. Soler
X, Ramsdell JW. Are asthma and COPD a continuum of
the same disease? J All Clin Immunol Pract. 2015; 489- 95. https://doi.org/10.1016/j.jaip.2015.05.030
89.
Ghebe M, Bafadhel M, Desai D, et al. Biological
clustering supports both ââDutchââ and ââBritishââ hypotheses of asthma and
chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;135:63-72. https://doi.org/10.1016/j.jaci.2014.06.035

| GalerĂa de imĂĄgenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|