

Autor : Esquivel Florencia1, Costantini Andrea2, Maspero Jorge3, Simón Gonzalo2, Moorat Anne, Mallett Steve, Carbone Ma. Florencia4, Penna Ma. Inés4
1 Laboratorios Phoenix SAICF. , 2GlaxoSmithKline Argentina, 3CIDEA Argentina, 4Laboratorios ELEA Phoenix S.A.
https://doi.org./10.56538/ramr.NNQC9291
Correspondencia : Florencia Esquivel E-mail: floresqui07@yahoo.com.ar
RESUMEN
El
asma es un grave problema de salud mundial. Según el último
informe del Ministerio de Salud, 1 380 000 sujetos padecen asma en la
Argentina.1 Las
guías internacionales (Europa, Estados Unidos, OMS) varían en su
enfoque para definir la equivalencia y la posibilidad de intercambio de los
productos para inhalación respiratorios. Si bien es posible un enfoque in
vitro, en general las guías recomiendan brindar más indiÂcios
clínicos que avalen la posibilidad de intercambiar el producto innovador
por otro posteriormente desarrollado con iguales principios activos en polvo
seco para inhalar. Este estudio, aleatorizado de fase IV, se realizó
para establecer la eficacia, seguridad y tolerabilidad de Neumoterol® 400 en
comparación con el producto medicinal de refeÂrencia Symbicort
forte budesonida/fumarato de formoterol 320/9 μg, indicados 2 veces al día en pacientes
asmáticos. Además, se evaluó la preferencia de los
pacientes por uno u otro dispositivo.
Se
demostró la no inferioridad de la formulación evaluada en
comparación con el proÂducto medicinal de referencia. El límite
inferior del IC del 95% para la diferencia entre los tratamientos fue mayor que
el margen predefinido de no inferioridad de –125 mL
(diferencia: 0,044 l [IC del 95%: –0,008 a 0,096]). Asimismo, se comprobaron
valores más altos para el AUC0-10h
del FEV1 y
un mayor cambio respecto del puntaje basal en la prueba de control del asma el
día 29 para las cápsulas de budesonida/fumarato de formoterol 400/12 μg. En un análisis exploratorio sobre la
preferencia de los pacientes por los dispositivos, una mayor proporción
de participantes expresaron su preferencia global por la cápsula de budesonida/fumarato de formoterol 400/12 μg. No se
informaÂron diferencias en la incidencia de AE o SAE (del inglés Adverse
event: evento adverÂso y Severe
Adverse Event: evento adverso severo) graves durante
el tratamiento o después de este. El perfil de seguridad de ambos
productos en general concordó con el perfil comprobado de budesonida/fumarato de formoterol.
Palabras
clave: Asma;
Budesonida; Fumarato de formoterol; Dispositivo inhalatorio
ABSTRACT
Asthmais a serious worldwide health problem. According to the last report of the MinÂistry of Health, 1,380,000 subjects suffer from asthma
in Argentina.1
The International Guidelines
(Europe, United States, WHO [World Health Organization]) have varying approaches
to define the equivalence
and possibility of switching
respiratory inhalation products. Whereas an in vitro approach is possible, in general Guidelines recommend providing more clinical evidence that support
the possibility of switching from the innoÂvative product to another one subsequently developed with the same active ingredient in the form of dry powder
inhaler. This randomized, phase IV study has been conductÂed to establish the efficacy, safety and tolerability of Neumoterol® 400 compared to the reference medicinal product Symbicort forte, budenoside/formoterol fumarate 320/9 μg twice a day in asthmatic patients. Also, the patients’
preference for one device or
the other has been evaluated.
Theevaluated formulation has proven to be
non-inferior compared to the
reference medicinal product.
The lower 95% CI (confidence interval) limit for the
difference beÂtween treatments was higher than the
predefined non-inferiority margin of –125 mL (difference: 0.044 l [95% CI: –0.008 to 0.096]). Also, higher values
were evidenced for the AUC0-10h (are under the curve) of the FEV1 (forced expiratory volume in the first second)
and a more important change
of the baseline score in the asthma control test on day 29 for
the budenoside/formoterol fumarate capsules of
400/12 μg. In one exploratory test about the patients’ preference
for one device
or the other,
a higher proÂportion of participants expressed their global preference for the budenoside/formoterol fumarate capsule of
400/12 μg. No differences
were reported in the incidence of AEs (adverse events) or SAEs (serious
adverse events) during or after the
treatment. The safety profile of both products in general agreed with the verified
profile of budenoside/ formoterol fumarate.
Key
words: Asthma; Budenoside;
Formoterol fumarate; Inhalation device.
Recibido: 05/06/2021
Aceptado: 03/10/2022
INTRODUCCIÓN
El
asma es un trastorno inflamatorio crónico de las vías
aéreas, caracterizada por una hiperreactividad de la vía
aérea que produce episodios recurrentes de sibilancias, falta de aire,
opresión de pecho y tos, en especial, por la noche o a
la mañana temprano. El asma es una enfermedad de alta prevalencia
y representa un importante problema de salud pública.2, 3
Las
pautas de la Iniciativa Global para el Asma (GINA)3
destacan la necesidad de tratar la inflamación de las
vías aéreas en el asma, así como también de
reconocer la importancia de los medicamentos inhalados profilácticos,
como los corticosteroides inhalados (ICS) y las
combinaÂciones de ICS/b-agonistas adrenérgicos de acción
prolongada (LABA) (también denominados b-2 agonistas de acción
prolongada), como el producto que contiene la combinación de budesonida/fumaÂrato de formoterol (BFF).3
Symbicortforte® (con inhalador turbuhaler®)
fue autorizado en el año 2010 en la Argentina para tratar el asma y la
enfermedad pulmonar obstructiva crónica (EPOC).4
El
laboratorio Phoenix desarrolló Neumoterol® 400, una
combinación de dosis fija con BFF en polvo seco (DPI), en
cápsulas, a administrarse mediante un inhalador de dosis única
provisto por la empresa Plastiape. Esta
formulación consiste en una cápsula que contiene una
pequeña cantidad de polvo que comprende una mezcla de 400 μg de budesonida micronizada, 12 μg de fumarato de forÂmoterol micronizado y excipientes. La formulación
está indicada como terapia de mantenimiento para el asma y para tratar a
los pacientes que sufren de EPOC. Las guías internacionales de Europa,
los Estados Unidos y la OMS, entre otras, varían en cuanto al enfoque
para establecer la posibilidad de intercambio y la equivalencia de los
productos respiratorios para inhalación. Si bien es posible un enfoque in
vitro, en general las guías recomiendan brindar más indicios
clínicos que avalen la posiÂbilidad de intercambiar diferentes
formulaciones de iguales principios activos. No obstante, en las regiones con
mercados emergentes, el enfoque regulatorio de brindar licencias comerciales
para los inhaladores respiratorios en general se basa en establecer una
equivalencia farmacéutica solo a través de análisis in
vitro. En el caso de la Argentina, la cápsula de BFF (Neumoterol® 400) se
aprobó solamente sobre la base de indicios in vitro.
El
actual estudio de fase IV se realizó para demostrar la no inferioridad
(objetivo primario) y recoger indicios científicos en cuanto a la
eficacia, la seguridad y la tolerabilidad, así como también la
preferencia de los pacientes por la cápsula de BFF 400/12 μg (Neumoterol®) en
comparación con el producto medicinal de referencia (PMR) BFF 320/9 μg (Symbicort forte®)
en pacientes asmáticos.
MÉTODOS
Diseño del
estudio
Se
trató de un estudio de fase IV, multicéntrico,
abierto, aleatorizado, cruzado en dos sentidos, de no inferioridad para
comparar la eficacia, la seguÂridad y la tolerabilidad de Neumoterol® 400 (BFF 400/12
μg) administrado mediante su dispositivo inÂhalador
específico y el PMR, Symbicort forte® (BFF
320/9 μg) administrado mediante su dispositivo turbuhaler® en sujetos
asmáticos adultos. Figura 1.
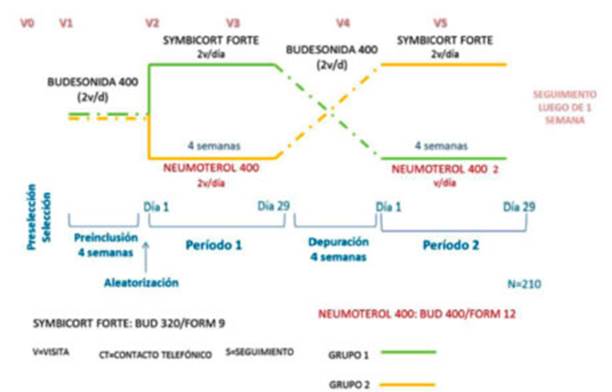
El
estudio se llevó a cabo de acuerdo con las Buenas Prácticas
Clínicas (GCP) y Conferencia internacional de armonización (ICH)
y todos los requisitos vigentes de privacidad del sujeto, además de los
principios éticos que se detallan en la Declaración de Helsinki
2008. Se obtuvo el consentimiento informado escrito de cada sujeto antes de
realizar cualquier procedimiento especíÂfico del estudio.
El
estudio consistió en seis fases: la preselección, la
selección/preinclusión (4 semanas), el
período de tratamiento 1 (4 semanas), el reposo farmacológico
(por lo menos, 4 semanas), el período de tratamiento 2 (4 semanas) y el
seguimiento (1 semana). La duraÂción total para cada sujeto fue de, al
menos, 17 seÂmanas. El cronograma incluyó hasta seis visitas del estudio
y un llamado telefónico de seguimiento.
En
la visita de preselección, se obtuvo el conÂsentimiento informado antes
de realizar cualquier evaluación y cambio en el régimen de
medicación de cada participante. En la preselección, se instruÂyó
a los pacientes acerca de la visita de selección y de preinclusión. En la visita 1 (visita de
selección y de preinclusión), los
sujetos que cumplieron con los criterios de inclusión ingresaron en un
período de preinclusión de 4 semanas.
La información sobre los criterios de inclusión y de
exclusión se detalla en las Tablas 1 y 2.


Durante
el período tanto de preinclusión como
de reposo farmacológico, todos los sujetos recibieÂron 400 μg de budesonida mediante el
DPI 2 Ă— d. Se permitió que todos los participantes usaran
medicación de rescate (inhalador con medidor de dosis presurizado de
salbutamol 100 μg) durante todo el
período del estudio y hasta la visita 5.
Al
final del período de preinclusión, los
pacientes volvieron a ser evaluados; se le indicó a cada sujeto que se autoadministrara el tratamiento del estudio en cada
período de tratamiento durante 4 semaÂnas de la siguiente manera,
según el cronograma de aleatorización: a) inhalación de
una cápsula de Neumoterol®
400 (BFF 400/12 μg) mediante su
dispositivo, 2 veces por día: una cápsula por la mañana y
otra por la tarde/noche (aproximaÂdamente 12 h después). b)
inhalación de PMR mediante turbuhaler® (BFF 320/9 μg) 2 veces por día cada mañana y cada
tarde (aproximadamente 12 h después). Luego de una fase de reposo farmaÂcológico
de 4 semanas, los participantes pasaron al tratamiento alternativo según
el cronograma de aleatorización. Se llevaron a cabo evaluaciones de
eficacia, farmacodinámica (PD), encuesta de
preferencia de los pacientes por los dispositivos y evaluaciones de seguridad.
Los
sujetos elegibles a fin de participar en el esÂtudio debían tener en la
visita 1 (visita de selección y de preinclusión), un mejor volumen espiratorio forzado
en el primer segundo (FEV1)
antes del bronÂcodilatador entre ≥ 40% al ≤ 85% del valor normal
previsto. El porcentaje previsto se calculó usando las ecuaciones de
referencia de la Iniciativa Global de la Función Pulmonar de la Sociedad
RespiratoÂria Europea5 y
aplicando ecuaciones o ajustes por etnia según correspondiera. La
reversibilidad de la enfermedad fue demostrada con la mejora del FEV1 (≥ 12% y ≥ 200 mL) en un lapso de 10 min a 40 min luego de 2 a 4
inhalaciones de aerosol para inhalar de salbutamol (o tratamiento de
nebulización equiÂvalente con solución de salbutamol). Asimismo,
el límite de estabilidad del FEV1 se
consideró un punto de referencia del estado de asma del sujeto en la preÂinclusión y se usó para la
comparación durante toda la fase de tratamiento a fin de evaluar la seguridad
del sujeto. Se calculó en la visita 2 como el 75% del mejor FEV1 antes del salbutamol.
Los sujetos eran retirados del estudio si no asisÂtían a la
clínica para realizar una visita requerida por el estudio y si no
regresaban para la visita reprogramada o no podían contactarse a fin de
reprogramar la visita omitida. Si se continuaba sin poder contactar al
participante, se consideraba que el sujeto se había retirado del estudio
con un motivo primario de “perdido en el seguimiento”. Además, los
sujetos se retiraban si se cumplía alÂguno de los siguientes criterios
luego de tener en cuenta la duración promedio del QTc
de electrocarÂdiogramas por triplicado: a) QTc >
500 ms; b) QT sin corregir > 600 ms; c) aumento de al menos 60 ms en
relación con el QTc basal.
No se permitían privilegios o exenciones al protocolo,
excepto por inquietudes de seguridad inmediatas.
Cumplimiento: Los sujetos recibieron el trataÂmiento del
estudio en su casa, excepto las dosis matinales de las visitas 2 a 5, que
recibieron en la clínica, y fueron observados por el personal del
estudio para garantizar una administración adecuada. El cumplimiento de
los participantes se evaluó en las visitas clínicas 3 y 5 y en la
discontiÂnuación prematura revisando el contador de dosis del dispositivo
del PMR y contando las cápsulas sin usar. Si la tasa de cumplimiento era
inferior al 80% o superaba el 120%, se volvía a educar al paciente con
respecto a la dosis indicada. Si el tratamiento se suspendía
prematuramente durante el estudio o si el cumplimiento estaba reiteradamente
fuera del rango aceptable, se contactaba al monitor del centro a fin de
analizar la elegibilidad del particiÂpante para continuar en el estudio.
El cumplimiento del tratamiento por parte del sujeto
también se evaluó mediante un contacto telefónico al final
de la segunda semana de cada período de tratamiento. El médico/el
personal del centro le había mostrado a cada sujeto cómo leer con
precisión los dispositivos antes de que comenÂzara el estudio.
Criterios de valoración: El criterio de
valoración primario de eficacia fue el cambio respecto del valor basal
en el FEV1 valle en la
mañana del día 29. Se definió como FEV1
valle al valor matinal antes del broncodilatador y antes de la
dosis, 12 h después de la última dosis vespertina (día
28), al concluir cada período de tratamiento.
Los criterios de valoración secundarios de eficacia
incluyeron el área bajo la curva (AUC) del FEV1
de 0-10 h al inicio de cada período de tratamiento (0
[antes de la dosis], 5 min, 15 min, 30 min; 1, 2, 5 y 10 h después de la
dosis matinal el día 1) y el cambio respecto del valor basal en el
puntaje de la prueba de control del asma (ACT)6 luego de las 4 semanas de cada
período de tratamiento. Se obtuvieron mediciones de espirometría
usando equipos que cumplieran con las recomendaciones mínimas de
rendimiento de la Sociedad Torácica EstadounidenÂse7 o que las superaran. Todos los
centros usaron su propio equipo de espirometría.
Se registró el FEV1 más
alto a partir de tres esfuerzos aceptables. Para determinar el AUC0-10 horas, se midió el
FEV1 en las visitas
clínicas 2 y 4, a los 0 (antes de la dosis), 5, 10 y 20 min; y a las 1,
2, 5 y 10 h después de la dosis matinal. La ACT fue un cuestionario
autocompleÂtado de 5 ítems desarrollado para medir el control del asma
del sujeto y que podía completarse con rapidez y facilidad en la
práctica clínica.6
La preferencia del sujeto por el dispositivo al final de cada
período de tratamiento se definió como un criterio de
valoración exploratorio. Dicha preferencia se analizó usando un
cuestionario. Durante la visita 3, se les pedía a los participantes que
completaran una encuesta de 3 preguntas; durante la visita 5 (fin del estudio),
se completaba una encuesta de 4 preguntas.
Los criterios de valoración de seguridad incluÂyeron el
cambio respecto del valor basal en los signos vitales (pulso y presión
arterial), el electroÂcardiograma y las evaluaciones de la bioquímica
clínica, la incidencia de eventos adversos (AE) durante cada
período de tratamiento, la incidenÂcia de exacerbaciones del asma
(definidas como el agravamiento del asma que exige un tratamiento que no sea el
del estudio o salbutamol de rescate, incluso la necesidad de corticoides
inhalados o sistémicos, o una visita a la guardia u hospitalización),
la incidencia de exacerbaciones graves del asma (definidas como el deterioro
del asma que requiere de corticosteroides
sistémicos durante al menos 3 días u hospitalización o
visita a la guardia a causa del asma que exigió corticoides
sistémicos), la incidencia de candidiasis oral evaluada mediante examen
y discontinuación prematura.
Análisis estadístico: Los
cálculos del tamaño de la muestra se basaron en el criterio de
valoración primario de eficacia. El cálculo de la variabilidad se
obtuvo a partir de un estudio anterior,9 en el que la desviación
estándar intrasujeto observada fue de 210 mL. Asumiendo ese valor, se requerirían 168 sujetos
para demostrar la no inferioridad del inhalador de Neumoterol®
400 (BFF 400/12 μg) y PMR con BFF
320/9 μg 2 veces por día, en adultos
asmáticos, teniendo en cuenta una diferencia verÂdadera de –50 mL con una potencia del 90% y un nivel de significancia
unilateral del 2,5%.
Se determinó –125 mL como margen
de no inferioridad de según las diferencias clínicas míÂnimas
importantes (MCID) para esta población de pacientes. Se demostró
anteriormente que las MCID para un rango de pacientes asmáticos era de
230 mL.9
A fin de considerar una tasa de abandono de 10% aproximadamente,
el número planificado de sujetos para aleatorizar
fue de 187 participantes. Debían seleccionarse alrededor de 234 sujetos
y se suponía una tasa de fracaso del 20% para poder llegar a 187 sujetos
aleatorizados y obtener 168 sujetos que completaran el estudio.
Se usó un análisis con intención de tratar
(ITT) para el análisis primario de eficacia. Se llevó a cabo un
análisis de eficacia de respaldo usando la población por
protocolo (PP). Se hicieron análisis de seguridad con la
población de seguridad. No se planeó un análisis interino
para este estudio.
El análisis primario de eficacia se realizó usando
un análisis de covarianza de efectos mixtos con el FEV1
basal, el grupo de tratamiento y el período como efectos
fijos, y el sujeto como coeficiente aleatorio. La no inferioridad se
evaluó examinando el límite inferior del intervalo de confianza
(0,025 nivel de significancia unilateral) comparándolo con el margen de
no inferioridad de –125 mL. Se hicieron otras
comparaciones entre el producto en estudio (BFF 400/12 μg)
administrado mediante un inhalador de cápsula y el PMR (BFF 320/9 μg) para los criterios de valoración
secundarios. Se consideró que dichas comparaciones brindaban respaldo, y
no se aplicaron ajuste de multiplicidad. Se usaron versiones actuales del software
SAS.
RESULTADOS
Datos basales
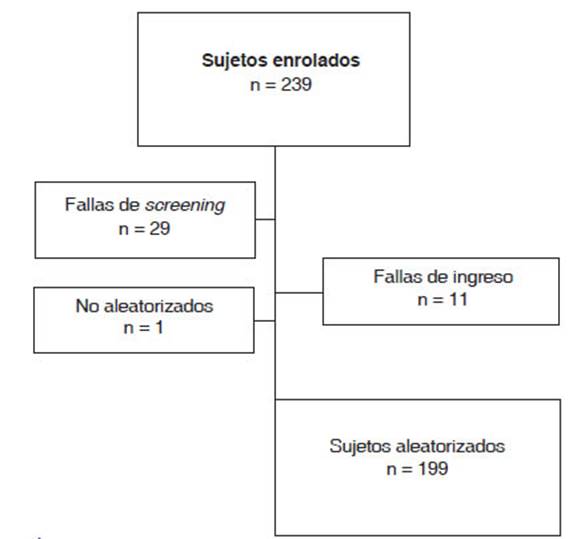
Se enrolaron en el estudio un total de 239 sujetos, y fueron
aleatorizados 199 sujetos (Figura 2). Entre los sujetos aleatorizados, 184
participantes (92%) completaron el estudio, y 15 sujetos (8%) se retiraron. Los
motivos frecuentes para retirarse fueron el retiro del consentimiento (n =
6; 3%) y desviaciones respecto del protocolo del estudio (n = 4; 2%).
Ningún sujeto se retiró por falta de eficacia. La
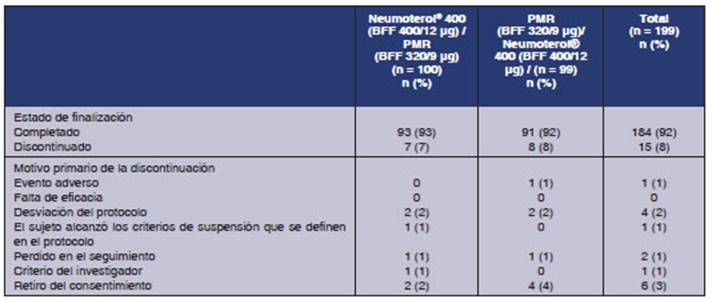
disposición de los sujetos según el período de tratamiento
se resume en la Tabla 3.
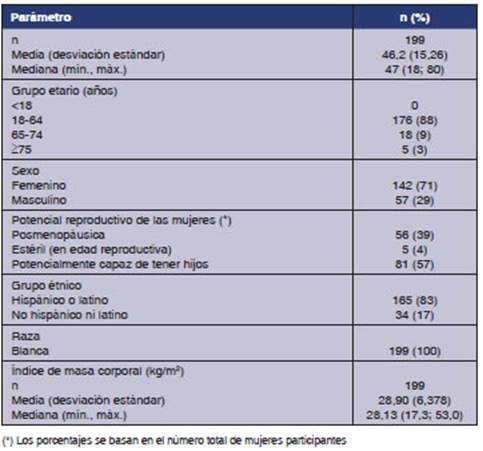
La demografía se aplica a ambos grupos debido al
diseño cruzado del estudio. La mediana de edad de los participantes fue
de 47 años, y la mayoría de los sujetos fueron mujeres (71%). Se
brinda una descripción completa de las características basales en
la Tabla 4.
En la visita de selección, la media del FEV1
antes del broncodilatador fue de 1,922 L (66,31% del vaÂlor
normal previsto), y la media del FEV1 posterior
al broncodilatador fue de 2,393 L (82,56% del valor normal previsto). La media
de la reversibilidad del FEV1 fue
de 470,60 mL (25,38%).
Se informaron factores de riesgo cardiovascular en 49 sujetos
(25%), incluidas hipertensión (n = 43; 22%), hiperlipidemia (n
= 10; 5%) y diabetes (n = 9; 5%). Durante el estudio, 106
participantes (53%) recibieron un medicamento concomitante o más. Los
que se recetaron con más frecuencia fueÂron los analgésicos, los
fármacos antihipertensivos y los antihistamínicos.
El cumplimiento del tratamiento fue de al menos el 80% para la
mayoría de los participanÂtes (188/191 sujetos [98,4%] para la
cápsula de Neumoterol® 400 y 183/187 sujetos [97,8%]
para el PMR. La mediana de cumplimiento estuvo en el rango del 94%-97% para ambas
formulaciones durante cada período de tratamiento.
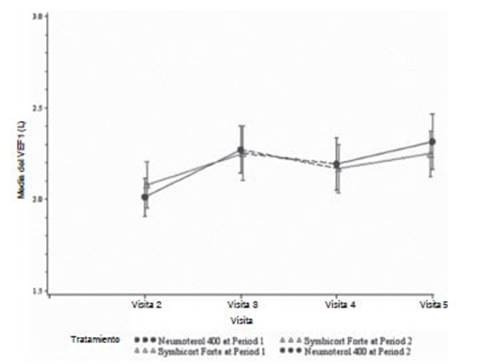
Resultados de eficacia: El criterio de valoración
primario de eficacia fue el cambio respecto del valor basal en el FEV1 valle en la mañana del
día 29. Para ambos tratamientos, hubo un aumento del FEV1
valle matinal en la población ITT. La media del aumento de
los mínimos cuadrados (LS) ajustados al modelo fue de 0,194L para Neumoterol® 400 y de 0,150 L para el PMR.
Quedó demostrada la no inferioridad de la cápsula de Neumoterol® 400 (BFF 400/12 μg): el límite inferior del intervalo de confianza (IC) del 95% para la diferencia de trataÂmiento
fue mayor que el margen de no inferioridad predeterminado de –125 mL (diferencia de 0,044 L; IC del 95%: –0,008; 0,096)
(Figura 3).
Un análisis realizado en la población PP concorÂdó
con el análisis primario en la población ITT. La no inferioridad
de la cápsula de Neumoterol® 400 (BFF 400/12 μg) quedó demostrada al compararla con el PMR
(BFF 320/9 μg) (diferencia de trataÂmiento:
0,043 l; IC del 95%: –0,012; 0,098).
Para ambos tratamientos, hubo una mejora en el FEV1
valle desde el período basal hasta el día 29 del
período 1 (de la visita 2 a la visita 3) y desde el período basal
hasta el día 29 del período 2 (de la visita 4 a la visita 5). Si
bien el FEV1 valle desÂcendió
durante la fase de reposo farmacológico de 4 semanas entre los
períodos 1 y 2, permaneció por encima del valor basal antes del
tratamiento (es decir, la media del FEV1 valle
en el período basal para el período 2 [visita 4] fue apenas mayor
que el nivel observado en el período basal para el período 1
[visita 2], independientemente del tratamiento).
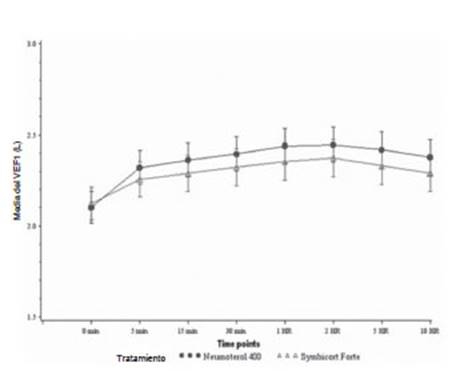
El AUC0-10 horas del
FEV1 al comienzo de cada
período de tratamiento fue un criterio de valoraÂción secundario
de eficacia. En la población ITT, el AUC0-10
horas del FEV1 el
Día 1 fue 0,98 L*h (IC del 95%: 0,576; 1,384) más alto para Neumoterol® 400 (BFF 400/12 μg) (Figura 4).
En la población ITT, ambos tratamientos se asociaron con un
aumento desde el período basal hasta el día 29 en el puntaje de
la ACT (otro criterio de valoración secundario de eficacia). La media
del aumento en los LS ajustada al modelo fue de 1,6 puntos para Neumoterol® 400 (BFF 400/12 μg) y de 1,0 punto para el PMR (BFF 320/9 μg). Esta diferencia de tratamiento favoreció a
la cápsula de BFF 400/12 μg, al haber una
diferencia de 0,6 puntos (IC del 95%: 0,1; 1,1).
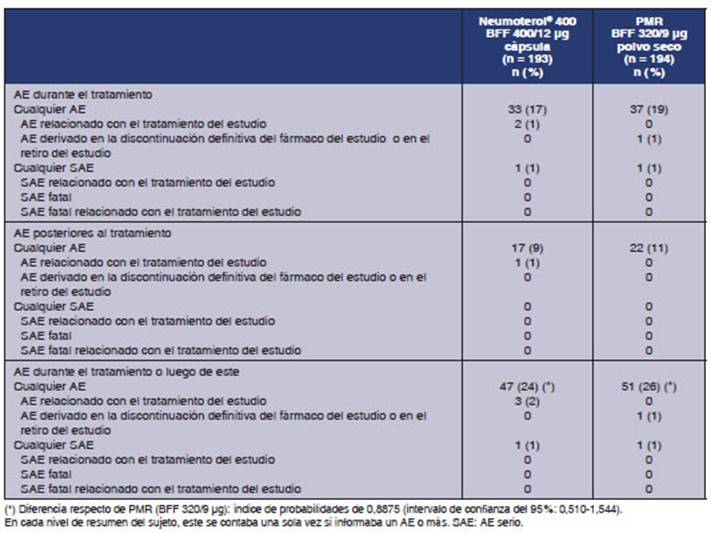
Resultados de seguridad: Se informaron AE durante el
tratamiento en 33 pacientes (17%) con la cápsula de BFF 400/12 μg y 37 sujetos (19%) que recibieron BFF 320/9 μg. Se informaron AE posteriores al tratamiento en 17
(9%) y en 22 participantes (11%), respectivamente. Se informó cualquier
AE durante el tratamiento o con posteÂrioridad a este en 47 sujetos (24%) para
la cápsula de BFF 400/12 μg y en 51
pacientes (26%) para BFF 320/9 μg. No hubo
diferencia estadísticamente significativa en cuanto a la incidencia de
al menos un AE durante el tratamiento o luego de este, entre ambos grupos de
tratamiento (índice de probabiliÂdades: 0,8875; IC del 95%: 0,510;
1,544) (Tabla 5). No se informaron muertes y ninguna participante quedó
embarazada durante el estudio.
Se informó un AE serio durante el tratamiento para la
cápsula de BFF 400/12 μg (colelitiasis),
y se informó un AE serio durante el tratamiento para BFF 320/9 μg (erupción) que llevó a la disÂcontinuación
definitiva del fármaco del estudio). El investigador no consideró
que ninguno de estos AE serios estuviera relacionado con el fármaco del
estudio. No se informaron SAE posteriores al trataÂmiento. Ningún sujeto
interrumpió el tratamiento ni fue discontinuado del estudio a causa de
un AE durante el tratamiento o después de este con la cápsula de
BFF 400/12 μg. Los AE informados con más
frecuencia durante el tratamiento se resumen en la Tabla 6. Los AE que se
informaron en, al menos, 2% de los participantes de cualquier grupo fueron
dolor de cabeza (4% y 3%, respectivamente), rinitis (2% y 3%) y bronquitis (1%
y 3%).

Los AE informados con más frecuencia con posÂterioridad al
tratamiento fueron infecciones e inÂfestaciones (4% para Neumoterol®
400 (BFF 400/12 μg) y 5% para el
PMR (BFF 320/9 μg)), trastornos del sistema
nervioso (1% y 4%, respectivamente), trastornos gastrointestinales (2% y 2%), y
trasÂtornos osteomusculares y del tejido conectivo (2% y 0%). El dolor de
cabeza representó el único AE posterior al tratamiento que
informaron al menos el 2% de los sujetos de cualquier grupo (2% para BFF 400/12
μg y 0% para BFF 320/9 μg).
El investigador consideró que dos AE durante el tratamiento
(palpitaciones y dolor de cabeza) y 1 AE posterior al tratamiento (aumento de
la creatina fosfoquinasa en sangre) estuvieron reÂlacionados
con la cápsula de BFF 400/12 μg. Para BFF
320/9 μg ninguno de los AE durante el traÂtamiento
o luego de este se consideró relacionado con el producto.
Dos sujetos (1%) presentaron una exacerbación moderada del
asma durante el tratamiento con BFF 400/12 μg.
Un participante (1%) tuvo una exacerbación grave del asma durante el
trataÂmiento en el tratamiento con BFF 320/9 μg.
Todas estas exacerbaciones del asma se resolvieron luego de la
intervención médica. Un paciente presentó una
exacerbación moderada del asma después del tratamiento al concluir
el período de tratamiento con BFF 320/9 μg,
la cual se resolvió.
No se informaron diferencias clínicamente releÂvantes entre
ambos tratamientos en la tendencia central para los valores de
bioquímica clínica en el período basal o el día 29,
ni cambios desde el período basal hasta el día 29, incluidos los
niveles de glucosa y potasio. No se advirtieron diferencias entre ambos
tratamientos en cuanto a los cambios en la presión arterial o la
duración del intervalo QT desde antes de la dosis hasta 30 min después
de la dosis el día 29.
Hubo un pequeño incremento de la frecuencia cardíaca
desde antes de la dosis hasta 10 min después de la dosis el día
29 para BFF 400/12 μg (media del aumento de los
LS de 1,3 latidos/min), mientras que la frecuencia cardíaca para BFF
320/9 μg permaneció esencialmente
invariable. Esta diferencia entre tratamientos para el cambio en la frecuencia
cardíaca fue estadísticamente sigÂnificativa (diferencia: 1,2
latidos/min; IC del 95%: 0,1; 2,3). La diferencia fue transitoria, y no hubo
diferencias entre la cápsula de BFF 400/12 μg
y BFF 320/9 para el cambio en la frecuencia cardíaca desde antes de la
dosis hasta 30 min después de la dosis el día 29 (diferencia: 0,2
latidos/minutos; IC del 95% –0,7; 1,2). No se informaron eventos de taquicardia
durante el estudio, pero un partiÂcipante tuvo un AE de palpitaciones leves 3
días después de iniciar el tratamiento con la cápsula de
BFF 400/12 μg. No hubo diferencias entre los dos
tratamientos para el cambio en la presión arterial sistólica o
diastólica desde antes de la dosis hasta 30 min después de la
dosis el día 29.
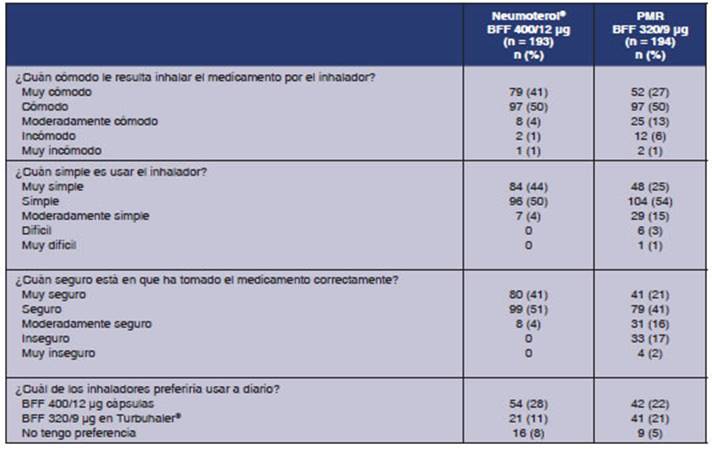
Criterio de valoración exploratorio: La prefeÂrencia
de los sujetos por los dispositivos al final de cada período de
tratamiento se definió como un criterio de valoración exploratorio.
Una proporción más alta de sujetos informaron que Neumoterol® 400 (BFF 400/12 μg) era «muy cómodo de usar» en
comparación con el PMR (BFF 320/9 μg),
(41% frente al 27%, respectivamente), “muy simple de usar” (44% frente al 25%),
y estuvieron muy seguros de haber usado el medicamento satisfacÂtoriamente (41%
frente al 21%). En general, más participantes manifestaron preferencia
por el inhalador de Neumoterol® 400 (BFF 400/12 μg), 50% frente al 32%. Se dispone de una
descripción detallada en la Tabla 7.
DISCUSIÓN
El asma es un grave problema de salud mundial. Su prevalencia está
en alza en muchos países, en espeÂcial, entre los grupos
pediátricos. Esta enfermedad les impone una carga inaceptable a los
sistemas sanitarios y la pérdida de la productividad laboral.2
En este estudio de fase IV, quedó demostrada la no
inferioridad de la eficacia de un inhalador con cápsula de BFF 400/12 μg (Neumoterol®
400) en comparación con el PMR (BFF 320/9 μg) adminisÂtrados 2 veces al día en adultos
asmáticos. BFF 400/12 μg (Neumoterol® 400) demostró mejoras en
los parámetros de la función pulmonar y el control de los
síntomas al final del período de tratamiento de 4 semanas.
Es importante destacar que no se informó una diferencia
entre ambos tratamientos en cuanto a la incidencia de AE o de AE serios durante
el traÂtamiento o después de este. En general, el perfil de seguridad de
ambas estrategias de tratamiento fue similar al que se informó antes
para BFF.10 Se informaron dos AE serios,
pero ninguno de los dos se consideró relacionado con el fármaco
del estudio.
No se informó una diferencia entre ambas estraÂtegias de
tratamiento en cuanto al cambio desde el período basal hasta el
día 29 en la frecuencia cardíaca antes de la dosis, la
presión arterial antes de la dosis, el QTc
antes de la dosis, los niveles de glucosa o los niveles de potasio.
Es interesante destacar que una evaluación exÂploratoria de
la preferencia de los participantes por los dispositivos reveló que una
mayor proporción de sujetos manifestaron una preferencia global por la
cápsula de BFF 400/12 μg en
comparación con la inhalación del PMR con BFF 320/9 μg (50% frente al 32%, respectivamente). Pocos
estudios examinaron las preferencias de los pacientes con diagnóstico de
asma o EPOC11 respecto a los disÂpositivos
para inhalación. Este aspecto representa un factor clave a fin de
mejorar el cumplimiento del tratamiento.
Este estudio tiene algunas limitaciones. Si bien podría
considerarse una debilidad del presente que sea de diseño abierto,
realizar un estudio ciego para medicamentos de vía inhalatoria en los
cuales el dispositivo no es intercambiable, no es factible. Por este motivo
para mejorar la sensibilidad del estudio, se realizó un diseño
cruzado en lugar de paralelo. Así mismo la modalidad abierta permiÂtió
adicionalmente la valoración exploratoria de preferencia del paciente
por el tipo de dispositivo.
Otras limitaciones son la aleatorización por centros y la
realización de espirometrías realizadas
con equipos comparables pero distintos en cada centro y no centralizadas.
CONCLUSIONES
Quedó
demostrada la no inferioridad de la cápsula de Neumoterol®
400 (BFF 400/12 μg) evaluada en
pacientes adultos asmáticos en comparación con el PMR (BFF 320/9 μg). Se observó una tendencia favorable con BFF
400/12 en la mejora en la funÂción pulmonar el 1° día (AUC 0-10
FEV1) y en el control de los
síntomas (ACT) del día 29.
Ambas formulaciones fueron bien toleradas y tuvieron un perfil de
seguridad congruente con las investigaciones anteriores. No se informaron informaron dos AE serios, pero ninguno de los dos se
consideró relacionado con el fármaco del estudio.
No se informó una diferencia entre ambas estraÂtegias de
tratamiento en cuanto al cambio desde el período basal hasta el
día 29 en la frecuencia cardíaca antes de la dosis, la
presión arterial antes de la dosis, el QTc
antes de la dosis, los niveles de glucosa o los niveles de potasio.
Es interesante destacar que una evaluación exÂploratoria de
la preferencia de los participantes por los dispositivos reveló que una
mayor proporción de sujetos manifestaron una preferencia global por la
cápsula de BFF 400/12 μg en
comparación con la inhalación del PMR con BFF 320/9 μg (50% frente al 32%, respectivamente). Pocos
estudios examinaron las preferencias de los pacientes con diagnóstico de
asma o EPOC11 respecto a los disÂpositivos para
inhalación. Este aspecto representa un factor clave a fin de mejorar el
cumplimiento del tratamiento.
Este estudio tiene algunas limitaciones. Si bien podría
considerarse una debilidad del presente que sea de diseño abierto,
realizar un estudio ciego para medicamentos de vía inhalatoria en los
cuales el dispositivo no es intercambiable, no es factible. Por este motivo
para mejorar la sensibilidad del estudio, se realizó un diseño
cruzado en lugar de paralelo. Así mismo la modalidad abierta permiÂtió
adicionalmente la valoración exploratoria de preferencia del paciente
por el tipo de dispositivo.
Otras limitaciones son la aleatorización por centros y la
realización de espirometrías realizadas
con equipos comparables pero distintos en cada centro y no centralizadas.
BIBLIOGRAFÍA
1.
Sergio J. Arias et al. Prevalencia y características clínicas del
asma en adultos jóvenes en zonas urbanas de Argentina. Arch Bronconeumol.2018;54(3):134-139.
2.
Mallol J, Solé D, Asher
I, et al. Prevalence of asthma
symptoms in Latin America: The International Study of Asthma and Allergies in Childhood (ISAAC). Pediatr Pulmonol. 2000;30:439-44.
https://doi.org/10.1002/1099-0496(200012)30:6<439::AID-PPUL1>3.0.CO;2-E
3.
Global Initiative for Asthma (GINA). Gina Report,
Global Strategy for Asthma Management and Prevention,
2016. In: www.ginasthma.org
4.
Symbicort Turbohaler
400/12, Inhalation powder.In:
https://www.medicines.org.uk/emc/medicine/11882 AcÂcessed:
September 20th, 2017.
5.
Quanjer PH, Stanojevic S,
Cole TJ, et al. Multi-ethnic referÂence
values for spirometry for the 3-95-yr age range: the globÂal lung function 2012 equations. Eur Respir J. 2012;40:1324- 43.
https://doi.org/10.1183/09031936.00080312
6.
Nathan RA, Sorkness CA, Kosinski M, et al. DevelopÂment
of the asthma control test:
a survey for assessing asthma control. J Allergy Clin Immunol.
2004;113:59-65.
https://doi.org/10.1016/j.jaci.2003.09.008
7.
Miller MR, Hankinson J, Brusasco
V, et al; ATS/ERS Task Force.
Standardisation of spirometry.
Eur Respir J. 2005;26:319-38. https://doi.org/10.1183/09031936.05.0003
4805
8.
GlaxoSmithKline. GSK Clinical Study
Register. In:
http://www.gsk-clinicalstudyregister.com/study/112202#ps AcÂcessed:
Sep 21st, 2017.
9.
Santanello NC, Zhang J, Seidenberg
B, et al. What are minimal important changes for asthma measures
in a clinical trial? Eur Respir J. 1999;14:23-7
https://doi.org/10.1034/j.1399-3003.1999.14a06.x
10.
Buhl R. Budesonide/formoterol for the treatment of asthma. Expert Opin Pharmacother. 2003;4:1393-406. https://doi.org/10.1517/14656566.4.8.1393
11.
Bereza BG, Troelsgaard Nielsen A, Valgardsson S, et al. Patient preferences in severe COPD and asthma: a comÂprehensive literature review. Int J Chron
Obstruct Pulmon Dis. 2015;10:739-44. https://doi.org/10.2147/COPD.S82179

| GalerĂa de imágenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|