

Autor : Directores: Asociación Argentina de Medicina Respiratoria Juan Antonio Mazzei , Jorge Osvaldo Cáneva
Autores designados por las siguientes instituciones
Asociación Argentina de Medicina Respiratoria
Bosio Martín Fernando
Casas Juan Pablo
Ciruzzi Julián
Ossés Juan Manuel
Schönfeld Daniel
Federación Argentina de Cardiología
Echazarreta Diego Federico
Perna Eduardo Roque
Sociedad Argentina de Cardiología
Diez Mirta
Vulcano Norberto Oscar
Sociedad Argentina de Pediatría
Giubergia Verónica
Haag Dora Fabiana
Sociedad Argentina de Reumatología
Laborde Hugo
Nitsche Alejandro
Autores invitados
Asociación Argentina de Medicina Respiratoria
Abouzed Roberto
Barro Analía
Bertorello Fancisco Andrés
Bustos Daniela Claudia
Chertcoff Julio Felipe
Coronel María Lorena
Diez Ana Rosa
Echazarreta Andrés
Enghelmayer Juan Ignacio
Figueroa Casas Marcelo
García González Jorge Andrés
Gómez Carmen Beatriz
Malamud Patricia Nora
Maldonado Lorena Vanesa
Manti Ariel Rolando
Mazzei Mariano
Morales Rosana
Naval Norma María
Papucci Tulio
Parpaglione Carlos
Peña David Armando
Picone Alejandro
Romera Andrés
Sabas Mario Fernando
Sheridan Lucas
Silio Julio Sergio
Svetliza Graciela
Tabaj Gabriela
Uribe Echevarría María Elisa
Federación Argentina de Cardiología
Piñeiro Daniel José
Sociedad Argentina de Cardiología
Amuchástegui Marcos
Atamañuk Andrés Nicolás
Belziti César
Bertolotti Alejandro Mario
Bortman Guillermo
Boughen Roberto
Favaloro Liliana Ethel
Favaloro Roberto
Iglesias Ricardo
Melero Marcelo
Perrone Sergio
Stewart Harris Alejandro
Talavera María Luján
Trivi Marcelo Sergio
Ubaldini Jorge
Sociedad Argentina de Reumatología
Catoggio Luis
Pisoni Cecilia
Scali Juan José
Academia Nacional de Medicina
Miguel de Tezanos Pintos
Ministerio de Salud de la Nación
María Laura Martínez
Facultad de Medicina - Universidad de Buenos Aires
Marcelo Melero
Revisores externos
Asociación Argentina de Medicina Respiratoria
Morandi Valeria
Schottlender Juan
Federación Argentina de Cardiología
Diez Fabián
Gregoretti Vanesa
Sociedad Argentina de Cardiología
Acosta Adriana
Lescano Adrián
Sociedad Argentina de Pediatría
D’Alessandro Virginia
Sociedad Argentina de Reumatología
Casado Gustavo
Sumario
PRÓLOGO
1. INTRODUCCIÓN, DEFINICIÓN, CLASIFICACIÓN, EPIDEMIOLOGÍA Y GENÉTICA................................................................................... 272
1.1 Introducción ........................................................................................................................................................................................ 272
1.2 Definición ............................................................................................................................................................................................ 273
1.3 Clasificación clínica.............................................................................................................................................................................. 274
1.4 Epidemiología y Genética.................................................................................................................................................................... 275
1.4.1 Epidemiología internacional....................................................................................................................................................... 275
1.4.1.1 Sexo-Edad................................................................................................................................................................... 276
1.4.1.2 Enfermedades del tejido conectivo.............................................................................................................................. 277
1.4.1.3 Hepatopatías crónicas................................................................................................................................................. 277
1.4.1.4 Infección por virus de inmunodeficiencia humana....................................................................................................... 277
1.4.1.5 Anorexígenos.............................................................................................................................................................. 277
1.4.1.6 Cardiopatías congénitas.............................................................................................................................................. 278
1.4.2 Epidemiología en Argentina....................................................................................................................................................... 278
1.4.3 Factores genéticos y hereditarios.............................................................................................................................................. 279
2. DIAGNÓSTICO ............................................................................................................................................................................................... 279
2.1 Clínica ................................................................................................................................................................................................. 279
2.2 Electrocardiograma.............................................................................................................................................................................. 280
2.3 Radiografía de tórax............................................................................................................................................................................. 281
2.4 Laboratorio .......................................................................................................................................................................................... 281
2.5 Ecocardiograma.................................................................................................................................................................................... 282
2.5.1 Ecocardiograma transesofágico................................................................................................................................................ 284
2.6 Laboratorio de Función Pulmonar...................................................................................................................................................... 284
2.7 Pruebas de esfuerzo............................................................................................................................................................................. 284
2.7.1 Prueba de marcha de seis minutos........................................................................................................................................... 284
2.7.2 Prueba de ejercicio cardiopulmonar.......................................................................................................................................... 285
2.8 Tomografía computada y angiotomografía........................................................................................................................................ 285
2.8.1 Tomografia axial computada (TAC) y tomografía axial computada de alta resolución (TACAR).............................................. 285
2.8.2 Angiotomografía con contraste.................................................................................................................................................. 285
2.9 Medicina nuclear................................................................................................................................................................................... 286
2.9.1 Centellograma ventilación/perfusión (V/Q)................................................................................................................................ 286
2.10 Angiografía pulmonar........................................................................................................................................................................... 286
2.11 Resonancia magnética nuclear........................................................................................................................................................... 286
2.12 Ultrasonografía ..................................................................................................................................................................................... 286
2.12.1 Ecografía Doppler abdominal ................................................................................................................................................... 286
2.12.2 Fibroscan o elastografía hepática ............................................................................................................................................. 286
2.13 Cateterismo cardíaco derecho ........................................................................................................................................................... 287
2.13.1 Prueba de sobrecarga con fluidos............................................................................................................................................. 288
2.13.2 Prueba de provocación por ejercicio.......................................................................................................................................... 288
2.14 Prueba de vasorreactividad pulmonar aguda.................................................................................................................................... 288
2.15 Cateterismo cardíaco izquierdo.......................................................................................................................................................... 289
2.16 Cinecoronariografía.............................................................................................................................................................................. 289
2.17 Pruebas genéticas................................................................................................................................................................................ 289
2.18 Biopsia pulmonar.................................................................................................................................................................................. 290
2.19 Algoritmo diagnóstico.......................................................................................................................................................................... 290
3. HIPERTENSIÓN ARTERIAL PULMONAR - Grupo 1.......................................................................................................................................... 292
3.1 Evaluación y pronóstico...................................................................................................................................................................... 292
3.1.1 Parámetros clínicos................................................................................................................................................................... 292
3.1.2 Capacidad funcional.................................................................................................................................................................. 293
3.1.3 Ecocardiograma......................................................................................................................................................................... 293
3.1.4 Resonancia magnética cardíaca................................................................................................................................................ 293
3.1.5 Parámetros hemodinámicos...................................................................................................................................................... 293
3.1.6 Capacidad de ejercicio............................................................................................................................................................... 293
3.1.6.1 Prueba de la distancia caminada en seis minutos...................................................................................................... 293
3.1.6.2 Pruebas de ejercicio cardiopulmonar.......................................................................................................................... 293
3.1.7 Biomarcadores........................................................................................................................................................................... 294
3.1.8 Otras evaluaciones.................................................................................................................................................................... 294
3.1.9 Consideraciones........................................................................................................................................................................ 294
3.2 Medidas terapéuticas generales.......................................................................................................................................................... 296
3.2.1 Actividad física y rehabilitación supervisada ............................................................................................................................. 296
3.2.2 Embarazo, control de la natalidad y terapia hormonal sustitutiva en la posmenopausia ......................................................... 296
3.2.3 Cirugía electiva.......................................................................................................................................................................... 296
3.2.4 Prevención de las infecciones .................................................................................................................................................. 297
3.2.5 Tratamiento de la anemia.......................................................................................................................................................... 297
3.2.6 Soporte psicosocial.................................................................................................................................................................... 297
3.2.7 Viajes en avión y zonas de altura.............................................................................................................................................. 297
3.3 Tratamiento farmacológico inespecífico............................................................................................................................................ 297
3.3.1 Anticoagulantes orales............................................................................................................................................................................ 297
3.3.2 Digoxina ..................................................................................................................................................................................... 297
3.3.3 Diuréticos................................................................................................................................................................................... 297
3.3.4 Oxigenoterapia........................................................................................................................................................................... 297
3.3.5 Otros ......................................................................................................................................................................................... 298
3.4 Terapia farmacológica específica....................................................................................................................................................... 298
3.4.1 Bloqueantes de los canales del calcio....................................................................................................................................... 298
3.4.2 Antagonistas de los receptores de la endotelina....................................................................................................................... 298
3.4.3 Inhibidores de la fosfodiesterasa-5............................................................................................................................................ 299
3.4.4 Estimulantes de la guanilato ciclasa.......................................................................................................................................... 299
3.4.5 Análogos de las prostaciclinas (prostanoides) y agonistas de los receptores de prostaciclinas............................................... 299
3.4.6 Monoterapia............................................................................................................................................................................... 300
3.4.7 Terapia combinada.................................................................................................................................................................... 300
3.5 Abordaje terapéutico de las complicaciones..................................................................................................................................... 302
3.5.1 Arritmias ..................................................................................................................................................................................... 302
3.5.2 Hemoptisis................................................................................................................................................................................. 302
3.5.3 Complicaciones mecánicas....................................................................................................................................................... 302
3.6. Manejo de la insuficiencia ventricular derecha avanzada................................................................................................................ 303
3.6.1 Internación en Unidad de Terapia Intensiva.............................................................................................................................. 303
3.6.2 Septostomía auricular con balón............................................................................................................................................... 303
3.7 Trasplante pulmonar y cardiopulmonar....................................................................................................................................................... 304
3.8 Algoritmo terapéutico........................................................................................................................................................................... 305
3.9 Ética y cuidados al final de la vida...................................................................................................................................................... 306
4. HIPERTENSIÓN ARTERIAL PULMONAR ESPECIFICA............................................................................................................................... 307
4.1 HAP en pediatría................................................................................................................................................................................... 307
4.1.1 Definición................................................................................................................................................................................... 307
4.1.2 Clasificación............................................................................................................................................................................... 307
4.1.3 Cardiopatías congénitas............................................................................................................................................................ 308
4.1.4 Diagnóstico................................................................................................................................................................................ 308
4.1.5 Seguimiento - Parámetros para el control del tratamiento......................................................................................................... 310
4.1.6 Tratamiento ............................................................................................................................................................................... 310
4.1.6.1 Bloqueantes cálcicos................................................................................................................................................... 310
4.1.6.2 Monoterapia................................................................................................................................................................. 310
4.1.6.3 Terapia combinada ..................................................................................................................................................... 311
4.1.6.4 Cirugía de descompresión del VD............................................................................................................................... 311
4.1.6.5 Trasplante pulmonar.................................................................................................................................................... 311
4.1.7 Consideraciones finales............................................................................................................................................................. 311
4.2 HAP asociada a enfermedades del tejido conectivo......................................................................................................................... 312
4.2.1 Consideraciones diagnósticas................................................................................................................................................... 312
4.2.2 Consideraciones terapéuticas.................................................................................................................................................... 312
4.3 HAP asociada a infección por el virus de la inmunodeficiencia humana....................................................................................... 313
4.3.1 Consideraciones diagnóstico terapéuticas................................................................................................................................ 313
4.4 HAP asociada con hipertensión portal (hipertensión portopulmonar)........................................................................................... 313
4.4.1 Consideraciones diagnósticas................................................................................................................................................... 313
4.4.2 Consideraciones terapéuticas.................................................................................................................................................... 314
4.5 HAP asociada con enfermedades cardiacas congénitas del adulto................................................................................................ 314
4.5.1 Consideraciones pronósticas..................................................................................................................................................... 314
4.5.2 Abordaje terapéutico.................................................................................................................................................................. 314
4.5.2.1 Manejo quirúrgico........................................................................................................................................................ 314
4.5.2.2 Abordaje farmacológico............................................................................................................................................... 315
4.6 HAP asociada con esquistosomiasis................................................................................................................................................. 315
4.7 Enfermedad pulmonar veno-oclusiva y hemangiomatosis capilar pulmonar - Grupo 1’.............................................................. 315
4.7.1 Consideraciones diagnósticas................................................................................................................................................... 316
4.7.2 Consideraciones terapéuticas.................................................................................................................................................... 316
5. Hipertensión pulmonar por enfermedad cardíaca izquierda – Grupo 2.................................................................................................... 316
5.1 Definición ........................................................................................................................................................................................... 316
5.2 Fisiopatología........................................................................................................................................................................................ 317
5.3 Diagnóstico ........................................................................................................................................................................................... 318
5.4 Tratamiento ........................................................................................................................................................................................... 318
6. Hipertensión pulmonar por enfermedad pulmonar y/o hipoxia – Grupo 3............................................................................................... 319
6.1 Definición .......................................................................................................................................................................................... 319
6.2 Diagnóstico .......................................................................................................................................................................................... 320
6.3 Tratamiento .......................................................................................................................................................................................... 320
7. HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA Y OTRAS OBSTRUCCIONES DE LA
ARTERIA PULMONAR – Grupo 4................................................................................................................................................................. 312
7.1 Diagnóstico ......................................................................................................................................................................................... 322
7.2 Tratamiento ......................................................................................................................................................................................... 322
7.2.1 Quirúrgico.................................................................................................................................................................................. 322
7.2.2 Terapia farmacológica............................................................................................................................................................... 323
8. HIPERTENSIÓN PULMONAR CON MECANISMOS MULTIFACTORIALES Y/O NO ESCLARECIDOS – GRUPO 5................................. 324
9. CENTRO DE REFERENCIA EN HIPERTENSIÓN PULMONAR.................................................................................................................... 325
9.1 Derivación al CRHP ............................................................................................................................................................................. 325
9.2 Recursos humanos............................................................................................................................................................................... 325
9.3 Requisitos de servicios permanentes................................................................................................................................................ 326
9.4 Estudios diagnósticos.......................................................................................................................................................................... 326
9.5 Red de interconexión con otros servicios con posibilidades de rápida derivación...................................................................... 326
9.6 Actividades académicas...................................................................................................................................................................... 326
10. COMENTARIOS FINALES Y CONCLUSIÓN.................................................................................................................................................. 326
11. RESUMEN DE ASPECTOS DESTACADOS................................................................................................................................................... 328
ABREVIATURAS Y ACRÓNIMOS
AD aurícula derecha
ALK-1 receptor tipo I del TGF-ß
ANA anticuerpos antinucleares
anti-nRNP anticuerpos antirribonucleoproteína nuclear
AR artritis reumatoidea
ARE antagonista receptor de la endotelina
BCC bloqueantes de canales de calcio
BMP proteína ósea morfogénica
BNP péptido natriurético cerebral
CC cardiopatía congénita
CCD cateterismo cardíaco derecho
CCI cateterismo cardiaco izquierdo
CF clase funcional
CPFE fibrosis pulmonar combinada con enfisema
CRHP centros de referencia en hipertensión pulmonar
DLCO capacidad de difusión pulmonar de monóxido de carbono
DLCOc capacidad de difusión pulmonar de monóxido de carbono corregida para el nivel de hemoglobina
EAP endarterectomía de la arteria pulmonar
ECG elecrocardiograma
EIDPP enfermedad intersticial difusa del parénquima pulmonar
EMTC enfermedad mixta del tejido conectivo
ENG endoglina
EPOC enfermedad pulmonar obstructiva crónica
EPVO enfermedad pulmonar venooclusiva pulmonar
ES esclerosis sistémica
ESC European Society of Cardiology
ESR European of Respiratory Society
ETC enfermedades del tejido conectivo
FAN factor antinuclear
FR factor reumatoideo
GC gasto cardiaco
GDTP gradiente diastólico transpulmonar
GPD gradiente de presión diastólica
GPTP gradiente de presión transpulmonar
HP hipertensión pulmonar
HAP hipertensión arterial pulmonar
HAPD hipertensión arterial pulmonar por drogas y toxinas
HAPH hipertensión arterial pulmonar hereditaria
HAPI hipertensión arterial pulmonar idiopática
HCP hemagiomatosis capilar pulmonar
HPoP hipertensión portopulmonar
HPpca hipertensión pulmonar poscapilar aislada
HPPRN hipertensión pulmonar persistente del recién nacido
HPTEC hipertensión pulmonar tromboembólica crónica
IC índice cardíaco
IPDE-5 inhibidor de la fosfodiesterasa-5
ISRS inhibidores selectivos de la recaptación de serotonina
LES lupus eritematoso sistémico
NT-proBNP propéptido natriurético cerebral N-terminal (NT-proBNP
NYHA New York Heart Association
OMS Organización Mundial de la Salud
ON óxido nítrico
PAP presión arterial pulmonar
PAPD presión arterial pulmonar diastólica
PAPE presión arterial pulmonar de enclavamiento
PAPm presión arterial pulmonar media
PAPS presión arterial pulmonar sistólica
PDAP presión diastólica arteria pulmonar
PECP prueba de ejercicio cardiopulmonar
PETCO2 presión end-tidal CO2
PFDVI presión de fin de diástole del ventrículo izquierdo
PM6M prueba de marcha de seis minutos
POAP presión de oclusión de la arteria pulmonar
PPs presión pulmonar sistólica
PVRPA prueba de vasorreactividad pulmonar aguda
RMN resonancia magnética nuclear
RVP resistencia vascular pulmonar
RVS resistencia vascular sistémica
SCEF síndrome combinado enfisema/fibrosis
SO2 saturación arterial de oxígeno
SvO2 saturación de oxigeno en sangre venosa mixta
TAC tomografía axial computarizada
TACAR tomografía axial computarizada de alta resolución
TAPSE excursión sistólica del anillo tricuspídeo
TGF-b factor de transformación del crecimiento beta
TJV velocidad del chorro tricuspídeo
UW unidades Wood
VE/VCO2 equivalente ventilatorio del dióxido de carbono
VD ventrículo derecho
VD/VT espacio muerto
VI ventrículo izquierdo
VIH virus de inmunodeficiencia humana
VM volumen minuto
VO2max consumo máximo de oxígeno
VO2AT umbral anaeróbico
VPRT velocidad pico de regurgitación tricuspídea
V/Q ventilación/perfusión
Prólogo
Estas Guías Argentinas de Diagnóstico y Tratamiento de la Hipertensión Pulmonar compendian y evalúan las evidencias aportadas por el IV Simposio Mundial de Hipertensión Pulmonar realizado en Dana Point, Estados Unidos, el V Simposio Mundial de Hipertensión Pulmonar realizado en Niza, Francia, y la reciente actualización que hicieran las sociedades científicas European Society of Cardiology (ESC) y European of Respiratory Society (ERS). El presente documento es una adaptación a nuestro medio de los lineamientos mencionados.
A la hora de interpretar esta información debe tenerse presente que se trata de una patología extremadamente infrecuente y que las recomendaciones emitidas se elaboraron en base a las evidencias disponibles al momento en que fueron escritas. Por lo cual pueden no ser las esperadas para los diversos abordajes propuestos los cuales, por otra parte, están en permanente revisión y cambio.
Es precisamente en estas situaciones que las Guías pueden ayudar al médico en su práctica diaria en la elección de la mejor conducta diagnóstico terapéutica para la Hipertensión Pulmonar a fin de revertir la alta morbilidad y mortalidad. Si bien se alienta al profesional a seguir las recomendaciones de Guías, éstas no anulan el criterio clínico y dejan la decisión final a cargo del médico tratante quien también basa su decisión en la condición clínica general del paciente, la opinión del propio paciente y su familia y las regulaciones vigentes relacionadas con las políticas de salud.
Los especialistas que han contribuido a la confección de estas Guías de Consenso y los revisores externos pertenecen y han sido designados por la Asociación Argentina de Medicina Respiratoria (AAMR), Sociedad Argentina de Cardiología (SAC), Federación Argentina de Cardiología (FAC), Sociedad Argentina de Pediatría (SAP) y Sociedad Argentina de Reumatología (SAR); y fueron invitados a participar por los Directores.
1. Introducción, definición, clasificación y epidemiología
1.1 Introducción
La hipertensión pulmonar (HP) abarca un grupo heterogéneo de entidades clínicas, con un espectro amplio de cambios patológicos a nivel vascular pulmonar que conducen a incrementos de la resistencia vascular pulmonar (RVP) y de la presión arterial pulmonar (PAP) que determinan finalmente el fallo ventricular derecho y la muerte. Esta entidad no es una enfermedad per se sino una condición hemodinámica que comparten múltiples etiologías. Puede aparecer en todas las décadas de la vida aunque predomina en la adultez, genera discapacidad a quien la padece y causa elevada mortalidad. Su diagnóstico es habitualmente tardío por lo que se debe tener una alta sospecha que permita implementar rápidamente los estudios y tratamientos adecuados a fin de evitar el deterioro clínico del paciente.
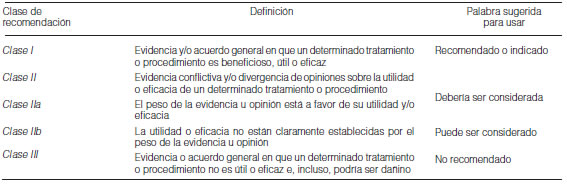
El Cuarto Simposio Mundial de Hipertensión Pulmonar de 2008 realizado en Dana Point, California1, 6 sirvió como marco para la publicación, en nuestro medio, del artículo de “Actualización en el diagnóstico y tratamiento de la hipertensión pulmonar”7. Más adelante, en el año 2013, con los antecedentes de la reunión de 2008 y nuevas evidencias, expertos reunidos en el Quinto Simposio Mundial de Hipertensión Pulmonar, realizado en Niza en el mes de marzo elaboraron un Consenso de Diagnóstico y Tratamiento para la Hipertensión Pulmonar proponiendo varios cambios, entre ellos modificaciones en la definición, clasificación y diagnóstico (imágenes) y en la descripción patobiológica. Se analizaron los efectos de nuevos fármacos y se incorporaron cambios al algoritmo de tratamiento y se definieron metas u objetivos terapéuticos basados en la evidencia de múltiples ensayos clínicos controlados. Asimismo, se incrementaron los conocimientos relacionados con los factores pronósticos y se estableció la relevancia de la relación diagnóstico temprano – tratamiento temprano; así como la del estudio y seguimiento de la función del ventrículo derecho (VD)1, 8. Estas nuevas Guías Argentinas de Consenso se basan, fundamentalmente, en dichos cambios considerando, además, los agregados en las Guías 2015 de Diagnóstico y Tratamiento de las sociedades ESC y ESR; a la vez que se proponen interrogantes acerca de temas en los cuales los conocimientos son aún insuficientes. A continuación, se consignan la Clase de recomendación y el Nivel de evidencia que se utilizarán en esta guía, cuando existiera (Tablas 1 y 2).

1.2 Definición
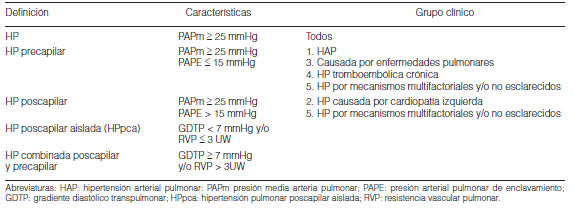
Se define hemodinámicamente a la HP como la elevación de la presión media de la arteria pulmonar (PAPm) con valores iguales o superiores a 25 mmHg registrados por cateterismo cardíaco derecho (CCD) y con el paciente en reposo9,10.
Por otra parte se define hipertensión arterial pulmonar (HAP) cuando a la definición anterior se le agregan una resistencia vascular pulmonar (RVP) mayor de tres Unidades Wood (UW) y una presión arterial pulmonar de enclavamiento (PAPE) menor o igual a 15 mmHg, en ausencia de otras causas de HP precapilar (enfermedades pulmonares, tromboembolismo crónico).
Consideraciones
– Esta definición, común a todas las formas de HP según las causas que la originen, puede presentarse con PAPE normal o elevada (Tabla 3).
– Se acepta como normal un valor de PAPm de hasta 20 mmHg. No hay suficientes datos para introducir el término HP borderline o limítrofe en pacientes con PAPm entre 21 y 24 mmHg, porque sus implicancias pronósticas y terapéuticas se desconocen. En estos pacientes está indicado un seguimiento estricto, particularmente cuando existe riesgo de desarrollar HAP (colagenopatías, antecedentes familiares de formas idiopática, hereditaria, infección virus de inmunodeficiencia humana [VIH], cirrosis hepática, etc.)9, 10.
– Los valores hemodinámicos normales durante el ejercicio no están bien definidos por lo que no se agregan criterios de diagnóstico relacionados con registros obtenidos en estas condiciones9. Se necesitan más estudios para definir qué niveles de elevación de la PAPm y de la RVP inducida por ejercicio tienen implicancias diagnósticas y terapéuticas. Los laboratorios funcionales demuestran valores dispares en la población sana sobre el comportamiento de presión pulmonar intraesfuerzo, por lo cual se ha desaconsejado su utilización en la definición. Sin embargo, existe suficiente evidencia sobre el valor pronóstico del incremento de los valores de presión pulmonar sistólica (PPs) con el ejercicio sobre el ventrículo derecho y en ciertas poblaciones especiales (esclerodermia, HAP hereditaria)11.
– Se debe evitar el uso de unidades internacionales para expresar la RVP, debiendo ser expresada en UW. El límite superior de RVP se mantiene en tres UW, pacientes con valores menores difícilmente tengan HAP9. La RVP no debe ser parte de la definición general de la HP, este parámetro se incluye en la definición hemodinámica de la HAP10.
– Para la caracterización de la HP combinada poscapilar y precapilar se adopta el gradiente diastólico transpulmonar (GDTP) de acuerdo al siguiente cálculo: PAPD (presión arterial pulmonar diastólica) – PAPE, considerando como normal un valor menor de 7 mmHg (Tabla 3)10.
– Se adoptan nueva terminología y subgrupos para la definición hemodinámica de la HP poscapilar10 (Tabla 3).
1.3 Clasificación clínica
Estas Guías Argentinas de Consenso adoptaron la clasificación de las Guías 2015 ESC/ESR las cuales, a su vez, modifican ligeramente la del Consenso de Niza 20139, 10 (Tabla 4)
A continuación se enumeran los cambios más prominentes:
– Se incluyeron condiciones frecuentemente halladas en niños
– Se incorporaron mutaciones recientemente identificadas
– La hipertensión pulmonar persistente del recién nacido pasó a ser subgrupo 1”
– Se agregó la obstrucción congénita/adquirida del tracto de entrada o salida del ventrículo izquierdo (VI), cardiomiopatías congénitas al Grupo 2.
– Se actualizó la clasificación de HAP asociada a enfermedades cardíacas congénitas (Tabla 5)
– La HP por neumopatía del desarrollo se incorporó al Grupo 3
– Se abandonó el término “HP desproporcionada” incorporándose el gradiente diastólico transpulmonar
– Se anexaron nuevos fármacos y drogas a los factores de riesgo de HP (Tabla 6)
– La HP precapilar asociada a anemia hemolítica crónica se desplazó del Grupo 1 al 5 (multifactorial y/o inespecífico)
– El subgrupo 1’ (enfermedad pulmonar veno-oclusiva (EPVO) y/o hemagiomatosis capilar pulmonar (HCP) se ha expandido
– El Grupo 4 se redenominó como HP por tromboembolismo crónico y otras obstrucciones de la arteria pulmonar
– La HP segmentaria se integró como un subgrupo al Grupo 5


1.4 Epidemiología y Genética
1.4 1 Epidemiología internacional
La HAP es una enfermedad rara. Los datos epidemiológicos provenientes de otros países demuestran una prevalencia de 5 a 25 casos/millón de habitantes y una incidencia de 1 a 2.4 casos/año/millón de habitantes12, 13, 14. El estudio escocés Scottish Pulmonary Vascular Unit, que considera aspectos de esta enfermedad desde una visión clínica en una institución dedicada al diagnóstico y tratamiento de la HAP halló una prevalencia de 52 casos/1.000.000 habitantes de 16 a 65 años de edad con una incidencia de 7.1 casos/1.000.000 habitantes/año(15). Es de destacar que a ninguno de estos pacientes se le había realizado un CCD. Sobre el particular se advierte que los datos epidemiológicos deben ser concretos y que, en el caso de este estudio, la HAP no fue confirmada por CCD tal y como lo exigen actualmente los lineamientos internacionales. El Registro Francés muestra una prevalencia de HAP confirmada por CCD de 15 casos/1.000.000 habitantes adultos y la incidencia de 2.4 casos/1.000.000 habitantes adultos/año. Se considera, internacionalmente, que los resultados de ambos estudios representan los rangos mayor y menor en cuanto a la prevalencia e incidencia de HAP12, 15. Aproximadamente la mitad de los pacientes con HAP padecen las formas idiopática (HAPI), hereditaria o inducida por drogas o fármacos (HAPD); en tanto las causas asociadas más frecuentes son las enfermedades del tejido conectivo, en particular la esclerosis sistémica12. La hipertensión arterial pulmonar familiar o hereditaria (HAPH) representa aproximadamente un 4-6% de los casos de HAP12, 13, 14. Sin embargo, se estima que esta prevalencia puede estar subestimada y podría ser más elevada. La transmisión es autosómica dominante con penetrancia incompleta, sin relación con el cromosoma X. Uno de los genes responsables denominado PPH1 está localizado en el locus 2q31/32/33/34 del cromosoma 216, 18. En estas familias nacen más mujeres que hombres19 hallazgo que podría expresar un mayor número de abortos en los embriones masculinos portadores del gen.
La prevalencia de HP del Grupo 2, asociada a enfermedades del corazón izquierdo, aumenta con el deterioro de la clase funcional, y con la gravedad de los síntomas en el caso de enfermedad valvular10. Respecto de la HP del Grupo 3, es más frecuente la HP leve en presencia de enfermedad intersticial y enfermedad pulmonar obstructiva crónica (EPOC) graves, en tanto que la HP grave se presenta en el síndrome combinado enfisema/fibrosis (SCEF)10. En el Registro Español la prevalencia e incidencia de hipertensión pulmonar por tromboembolismo crónico (HPTEC) fue de 3.2/1.000.000 y 0.9/1.000.000 por año, respectivamente20. Según el International CTEPH Registry el 74.8% de los pacientes con HPTEC tienen antecedentes de embolia pulmonar, siendo las condiciones asociadas más prevalentes: el síndrome de anticuerpos antifosfolipídicos, las deficiencias de proteínas S y C, la resistencia a la proteína C activada y la esplenectomía, entre otras21.
En lo que respecta a Sudamérica, Brasil aporta su experiencia con una cohorte de 178 nuevos casos de HAP confirmada por CCD estudiada en un centro de referencia entre 2008 y 2013. La edad promedio de la población examinada era de 46 años; predominaban las mujeres (3.3:1), el 45.5% pertenecía a la clase funcional NYHA III o IV. El 28.7% tenían HAPI, el 25.8% enfermedades del tejido conectivo y el 19.7% eran HAP asociada a esquistosomiasis. El 66% recibía tratamiento con inhibidores de la fosfodiesterasa-5 (IPDE-5), 27% con antagonistas de los receptores de la endotelina y el 5% con combinación de ambos. La supervivencia a los tres años alcanzó al 73.9%, con una diferencia significativa a favor de la HAPI y la HAP asociada a esquistosomiasis versus la HPTEC (p= 0.03)22. La relevancia de este estudio epidemiológico radica, fundamentalmente, en poner de relieve la participación de la esquistosomiasis como causa de HAP en la región, y las diferencias respecto de la información aportada por Estados Unidos y Europa.
Para concluir mencionamos otros datos sobre los cuales llaman la atención las Guías ESC/ERS como son: la ausencia de datos epidemiológicos comparativos entre grupos de HP; la escasa información demográfica y clínica sobre la HP de los Grupos 2 y 3; la carga que representan para la humanidad la HAP asociada a esquistosomiasis (p.ej.: Brasil) y la circunstancia de habitar por encima de los 2.500 metros sobre el nivel del mar10.
1.4.1.1 Sexo y edad
Las mujeres se ven afectadas dos veces más frecuentemente que los hombres y el riesgo de desarrollar la enfermedad es 1.7 veces mayor en los tres primeros meses después de un parto. Se ha reportado que la predominancia femenina desaparecería en los pacientes de más edad10. Recientemente, los resultados del Registro Francés modificaron el concepto de que la HAP era una enfermedad de mujeres jóvenes en la etapa fértil de la vida. La edad promedio de los pacientes incluidos en este
estudio fue 50 ± 15 años y un 10% de ellos tenía ≥ 75 años de edad. Este envejecimiento de la población afectada por la HAP también fue observado en un estudio realizado en la Mayo Clinic que demostró que uno de cada cuatro pacientes con esta dolencia tenía una edad ≥ 65 años23. Si bien es cierto que la HAP puede presentarse en cualquier edad, en los registros actuales aparece con mayor frecuencia entre los 50 y 65 años de edad10. En la infancia la distribución de la enfermedad por sexo es casi la misma14.
1.4.1.2 Enfermedades del tejido conectivo
La HAP asociada a enfermedades del tejido conectivo (ETC) constituye el 15.3% de los casos de HAP(12), con incidencias variables en función del método de diagnóstico utilizado. Un meta-análisis incluyó 12 estudios de HAP en esclerosis sistémica (ES), 3 estudios de HAP en lupus eritematoso sistémico (LES), uno de ellos también incluía pacientes con enfermedad mixta del tejido conectivo (EMTC) y 2 estudios de HAP en artritis reumatoide (AR)24. La prevalencia total de HAP fue del 13%, al igual que en aquellos con ES (18% en los 7 estudios con ecocardiograma Doppler y 8% en los 5 con confirmación por CCD)24. En los dos estudios de pacientes con LES la prevalencia de HAP confirmada por CCD fue de 3.24%; en AR (estudiada por ecocardiograma Doppler) fue de 22%. Algunas series informan prevalencias muy elevadas de HAP en pacientes con esclerodermia localizada, previamente conocida como Sindrome de CREST (calcinosis, Raynaud, esclerodactilia, esofagitis, telangiectasia), ES y ETC (5 al 75% de los casos diagnosticados)25. Un estudio en 384 pacientes con ES sin enfermedad respiratoria ni cardiopatía izquierda basal grave, seguidos durante una media de 41.03 ± 5.66 meses reveló una incidencia global de HP confirmado por CCD de 1.37/pacientes-año (IC95%% 0.74-2.00)26. La HAP es la causa más común de muerte en la esclerodermia27. Se han mejorado las técnicas de rastreo de HP en estos pacientes pero la supervivencia no ha aumentado mayormente lo que se debe, en parte, a las demoras en instaurar adecuados tratamientos y la necesidad de un manejo multidisciplinario.
1.4.1.3 Hepatopatías crónicas
El 2 a 10% de los pacientes con hepatopatías crónicas graves con hipertensión portal y hasta el 20% de los que candidatos a trasplante por cirrosis hepática tienen HAP28, 29. En las guías europeas se cita el trabajo de Krowka y cols. (2006) el cual comunica que desarrollan HAP el 1 a 2% de los paciente con enfermedad hepática e hipertensión portal y hasta el 5% de los candidatos a trasplante hepático. La distinción entre hipertensión porto pulmonar e HP en el marco de un estado hiperdinámico es de gran importancia. La HP hiperdinámica no representaría un factor de riesgo para la aparición de efectos adversos significativos durante la intervención y el seguimiento de un trasplante hepático30.
1.4.1.4 Virus de inmunodeficiencia humana
La HAP tiene una incidencia aproximada de 5.4 a 7.5% y una prevalencia de 0.46-0.57% en los pacientes con infección por VIH12, 13, 31, 32. Si bien es más común en los estadios avanzados de la infección, puede presentarse en cualquier momento evolutivo. El riesgo para desarrollar HAP es 2500 veces superior al de la población general y estaría relacionado con la presencia de los antígenos HLA-DR6 (subtipos DRB1*1301/2) y HLA-DR52 (subtipos DRB3*0301)31, 33. Algunos estudios han demostrado que la terapia antirretroviral de alta efectividad administrada en forma sostenida es capaz de modificar favorablemente las variables hemodinámicas de la HAP en estos enfermos34, 35; sin embargo este conocimiento es motivo de controversia.
1.4.1.5 Anorexígenos
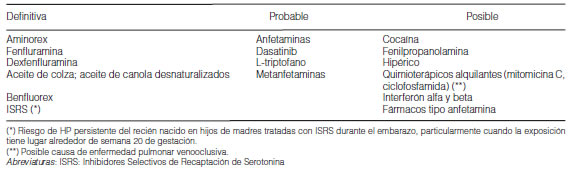
El consumo de anorexígenos fue un factor de riesgo significativo para el desarrollo de HAP en la década de 1960, cuando se describió la asociación de la HP primaria y el uso de fumarato de aminorex36. El 9.5% de las formas clínicas de HAP del Registro Francés estuvieron relacionadas con la exposición a estos fármacos12. El 24% de los pacientes que consumieron fenfluramina desarrollaron HAP en un periodo de 2 años, el 32% entre los 2 y 5 años y el 44% después de 5 años del tratamiento12.
1.4.1.6 Cardiopatías congénitas
La HAP es frecuente en los pacientes adultos con enfermedades cardíacas congénitas. En Europa y América del Norte la prevalencia de HAP asociada a cortocircuitos congénitos sistémico-pulmonares ha sido estimada entre 1,6 y 12,5 casos por millón de adultos, con un 25 a 50% de esta población afectada por el síndrome de Eisenmenger36. El Registro Francés demostró que las cardiopatías congénitas representaron el 11.3% de todas las formas clínicas de HAP12. El registro CONCOR (Holanda) reveló que el 4.2% de 5970 pacientes adultos con cardiopatías congénitas tenían HAP37.
1.4 2 Epidemiología en Argentina
En la Argentina se ha realizado más de un estudio epidemiológico con relevancia regional. El primero de ellos analizó todos los egresos hospitalarios identificados con el número de registro I27.0 (CIE10) con el diagnóstico básico de HAPI, registrados en la Dirección de Estadística e Información de Salud del Ministerio de Salud de la Nación de la República Argentina38. La información correspondiente al año 2005 da cuenta que durante ese año egresaron 160 pacientes con el diagnóstico de HAPI, el 43% tenía menos de 30 años de edad. Casi el 40% se concentró en la provincia de Buenos Aires. Al considerarse las defunciones se observó que en los años 2005 y 2006 fallecieron 115 pacientes con HAPI como causa básica de muerte; más del 50% de éstos eran mayores de 50 años; el 62,6% se concentraba en las provincias de Buenos Aires y Santa Fe y en la Ciudad Autónoma de Buenos Aires. Se atribuyen las diferencias en la concentración de casos a la localización de los centros especializados en la atención de esta enfermedad.
Más tarde, en 2014, se dieron a conocer los resultados del primer Registro Argentino de casos de HAP diagnosticados por CCD39. Se trata de 125 pacientes admitidos por primera vez en el consultorio de HP de la Fundación Favaloro, entre enero de 2004 y marzo de 2012. La población estudiada consistía en pacientes con HAP Grupo 1 (Dana Point 2008) confirmada por CCD. El 26.4% eran casos incidentales (síntomas iniciados en los 6 meses previos al CCD). La edad promedio era de 34 ± 15,7 años, con un claro predominio del sexo femenino 79,2% (3,8:1). El 48.8% tenía HAPI; el 28% HAP asociada a cardiopatías congénitas (n= 28 síndrome de Eisenmenger; n= 7 comunicación interauricular cerrada); el 14,4% tenía HAP asociada a ETC; el 6.4% HAP portopulmonar; el 1.6% HAP familiar y en el 0.8% (n= 1) de los casos se diagnosticó HAP asociada a infección por VIH. El seguimiento se extendió por 39.1 meses, al cabo de los cuales la supervivencia libre de trasplante fue de 63.2%; con tasas de supervivencia a los 12, 24 y 36 meses del 94%, 90% y 83%, respectivamente. En los pacientes pediátricos la supervivencia libre de trasplante fue del 62,5%. La supervivencia resultó mayor en los individuos con cardiopatías congénitas en comparación con el grupo de HAPI y HAP asociada a ETC. Las variables basales con asociación significativa a mortalidad y/o trasplante fueron la clase funcional III/IV, la distancia recorrida en la prueba de la caminata de 6 minutos < 380 metros, el derrame pericárdico, el deterioro significativo de la función sistólica del VD; y el índice cardíaco < 2.2 L/min/m2. Se destaca la similitud de sus resultados con otras series a nivel mundial.
El más reciente estudio publicado, comprendió la población total del país en el periodo 2000- 2009 (datos del Censo Nacional 2001 y proyecciones del INDEC) Nuevamente se incluyeron los fallecimientos identificados como I27.0 CIE10 en la base de datos de la Dirección de Estadística e Información de Salud del Ministerio de Salud, Presidencia de la Nación de la Republica Argentina40. La tasa anual de mortalidad por HAPI fue similar durante 10 años oscilando entre 1.39 y 2.39 muertes/1.000.000 habitantes (promedio 76 muertes/año); con un predominio en el sexo femenino (1.76 a 3.16/1.000.000) en comparación con los varones (0.9 a 2.11/1.000.000). La mortalidad más elevada se registró en los mayores de 70 años. Las tasas de mortalidad más altas se encontraron en las provincias de Tierra del Fuego (31.42/ 1.000.000) y San Juan (17.61/1.000.000); sin embargo, las tasas de mortalidad específica ajustadas por edad y sexo, en el año 2009, no muestran diferencias significativas entre las provincias. Para una población argentina de 40.091.359 habitantes, según el Censo Nacional 2010, se estima que habrá 601 a 2.085 casos prevalentes de HAP y 96 a 285 nuevos casos de HAP/año. Se ha recomendado implementar un Registro Nacional para disponer
de información más confiable sobre la morbilidad y mortalidad por HAPI y poder así promover la estandarización de las conductas diagnósticas y terapéuticas en todo el país, optimizando los recursos disponibles. Por el momento, está en marcha el Primer Registro Colaborativo de HP en Argentina (RECOPILAR) que mostrará el estado actual de la HAP nuestro país, ya que los datos epidemiológicos actuales no se correlacionan con las estimaciones. Los datos parciales disponibles no han sido publicados aún.
1.4.3 Factores genéticos y hereditarios
Grupo 1: La proteína ósea morfogénica (BMP por su sigla en inglés) es importante en la proliferación de las células vasculares41, 42. La mutación heterocigota del gen del receptor tipo II de la BMP (BMPR2) causa la muerte de algunas células y la proliferación de otras en el interior de las arterias pulmonares de pequeño calibre, siendo estas alteraciones responsables de los cambios histológicos de la HAP16, 17, 43. Se describe también una sobreexpresión de angiopoyetina-1 que interferiría con la función del gen produciendo cambios en el músculo liso y estimulando la angiogénesis. En el Grupo 1 estas mutaciones acontecen en el 75% de los casos de HAPH y el 25% de las HP esporádicas10. Se acepta actualmente que la mutación del BMPR2 es una condición necesaria pero no suficiente para el desarrollo de HP. La enfermedad genética tiene una penetración menor del 50%; sólo el 10% a 20% de los portadores de la mutación desarrollan HAP y existen ejemplos de mellizos idénticos con mutaciones del BMPR2, en los que sólo uno enferma44. Además, esta mutación se describe, también, en la HAP relacionada con anorexígenos45. Otras mutaciones, BMPR1B y SMAD9, sustentan el rol del Factor de Transformación del Crecimiento Beta (TGF-b) en la HAP10. En el 25% de las formas esporádicas de HCP y (EPVO) y, en el 100% de la HAPH frecuentes en familias consanguíneas se han observado mutaciones del gen EIF2AK4 codificador de una quinasa inductora de cambios en la expresión genética en respuesta a la deprivación de aminoácidos10. También se ha descubierto una mutación del receptor tipo I del TGF-b (ALK-1) o del receptor accesorio de la endoglina (ENG) que induce el desarrollo de la HP en el síndrome de telangiectasia hemorrágica hereditaria de Rendu-Weber-Osler44, 46, 47. En los pacientes con infección VIH y HAP no se ha encontrado el gen PPH148 y se presume que algunos mediadores inflamatorios podrían desencadenar los cambios histológicos.
Grupo 3: se sugiere que el polimorfismo contribuiría a la gravedad de la enfermedad en pacientes con EPOC e hipoxia10.
Grupos 2 y 4: no se han identificado mutaciones asociadas10.
Grupo 5: la heterogeneidad de este grupo no permite relacionarlo con factores genéticos10.
2. Diagnóstico
Para realizar el diagnóstico se debe seguir un procedimiento ordenado. Para estas Guías Argentinas de Consenso se ha tomado como base el algoritmo propuesto por la ESC/ERS7, 9, 10. Este proceso comienza con la sospecha clínica a partir de los síntomas, signos e historia sugestiva de HP y requiere la confirmación de los criterios hemodinámicos además de la identificación etiológica específica. Su abordaje es complejo y debe llevarse a cabo en instituciones especializadas9. Los programas de detección precoz (cribado) se aplican únicamente a pacientes de alto riesgo (p.ej.: enfermedades del tejido conectivo en el espectro de la ES, cardiopatías congénitas, infección por VIH, hepatopatías crónicas), antecedentes heredofamiliares, consumo de drogas o fármacos de probada influencia en la génesis de la enfermedad9.
2.1 Clínica
La mayoría de los signos y síntomas se relacionan con el deterioro de la función del VD7, 9, 10 (Tabla 7).

2.2 Electrocardiograma
Los cambios del electrocardiograma (ECG), cuando se observan, ya hablan de HP establecida. El ECG tiene una sensibilidad del 56% y una especificidad del 70% en la HP por lo que resulta inadecuado como prueba de detección de la enfermedad7, 49. Sin embargo, se debe señalar que su realización es imprescindible en la evaluación de un paciente con el diagnóstico confirmado. La taquicardia sinusal es la alteración del ritmo más frecuentemente encontrada, en tanto la arritmia supraventricular es un indicador de gravedad de la enfermedad (Tabla 8). Es importante tener en cuenta que un ECG normal no excluye el diagnóstico10 y la presencia de fibrilación auricular orienta hacia diagnósticos alternativos a la HAPI.

2.3 Radiografía de tórax
La radiografía de tórax presenta cambios en el 80% a 90% de los pacientes con HP en estadios avanzados50 (Tabla 9). No obstante, no existe correlación entre la gravedad de la HP y la magnitud de las alteraciones radiológicas y, una Rx normal no excluye el diagnóstico10.

2.4 Laboratorio
Se recomienda realizar en todos los pacientes exámenes de rutina; dosajes de hormonas tiroideas; serología para VIH, hepatitis B y C; y cribado básico para identificar ETC y hepatograma5, 9.
El cribado básico para ETC debería incluir factor antinuclear (FAN), antiDNA y factor reumatoideo (FR). Títulos de FAN mayores a 1/100 sugieren ETC. En pacientes con diagnóstico definido, ya sea de esclerodermia u otra ETC, es útil la determinación de otros autoanticuerpos (Tabla 10). El anticuerpo anticentrómero se expresa con más frecuencia en la variante limitada de esclerodermia que es la que más se asocia a HP. El anti-Scl-70 es más característico de la variante difusa y está más relacionado con la enfermedad pulmonar intersticial.
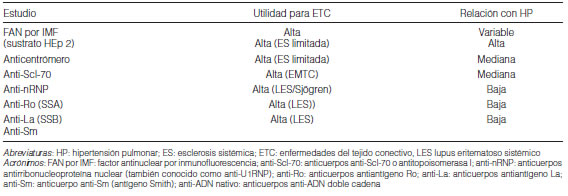
La presencia de anticuerpos antirribonucleoproteína nuclear (anti-nRNP) también conocido como anti-U1RNP, es característica de la enfermedad mixta del tejido conectivo (EMTC) y ocupa el segundo lugar en riesgo de asociarse con HP, después de la esclerodermia. Sin embargo, hay que considerar que se ha detectado la presencia FAN en el 40% de HAPI5, 51. El hallazgo de un FAN negativo hace poco probable el diagnóstico de LES pero no descarta otras ETC; vale tener presente que títulos elevados de FAN pueden observarse en pacientes con Enfermedad de Hashimoto. El LES puede asociarse a anticuerpos anticardiolipina10.
En pacientes con HPTEC se recomienda descartar trombofilia (títulos de anticoagulante lúpico, anticardiolipinas IgG e IgM, anti β2Glicoproteína IgG e IgM) y razón internacional normatizada (RIN) cuando esté en tratamiento con anticoagulantes5, 52, 53.
La concentración plasmática de ácido úrico, péptido natriurético cerebral (BNP)54 o del propéptido natriurético cerebral N-terminal (NT-proBNP), conjuntamente con el nivel de troponina55 están relacionados con la gravedad y pronóstico de la enfermedad56-61.
El BNP plasmático es un predictor independiente de mortalidad y la magnitud de su aumento se correlaciona positivamente con las cifras de PAPm, RVP y CF NYHA, y negativamente con los metros recorridos en la prueba de marcha de seis minutos, el volumen minuto (VM) y la supervivencia54. Al interpretar estos resultados es necesario tener en cuenta la existencia de factores que modifican estos valores, bien aumentándolos (esfuerzo, insuficiencia renal, enfermedad pulmonar, hipertensión arterial, hipertiroidismo, tratamiento con glucocorticoides, cirrosis hepática con ascitis o hemorragia subaracnoidea) o disminuyéndolos (obesidad, tratamiento con inhibidores de la enzima de conversión de angiotensina, bloqueadores beta, espironolactona y otros diuréticos62.
2.5 Ecocardiograma
La ecocardiografía es el paso inicial obligado para la evaluación de pacientes con sospecha de HP. En la valoración mediante eco-Doppler de la HP se pueden incluir tres objetivos diferentes: detección de valores elevados de la PAPm, evaluación funcional del VD y realización de un diagnóstico diferencial con el fin de detectar condiciones subyacentes, tales como cardiopatías congénitas, enfermedad valvular, enfermedad cardíaca izquierda, o trombos centrales responsables de la presencia de la enfermedad tromboembolica crónica. Es, además, útil para monitorear la progresión de la enfermedad en el tiempo y es una herramienta fundamental con valor pronóstico5, 9, 63. El ecocardiograma se debe realizar siempre que se sospeche HP y se puede usar para inferir el diagnóstico en pacientes cuyas múltiples mediciones sean consistentes con la enfermedad (Tabla 11)10. Para analizar la probabilidad ecocardiográfica de HP, se recomienda, en las nuevas Guías 2015, medir con Doppler de onda continua la velocidad pico de regurgitación tricuspídea (VPRT) en reposo (Tabla 11) y no utilizar la estimación de la presión pulmonar sistólica (PPs)10.
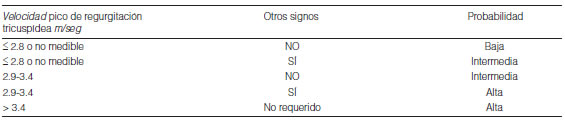
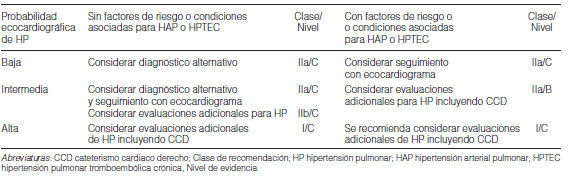
La presencia de variables de dos categorías diferentes (A/B/C) (Tabla 12) modifica la probabilidad ecocardiográfica de HP10; conocer la probabilidad de desarrollar HP a futuro o padecerla en el momento del examen permite implementar estrategias adicionales de diagnóstico y/o seguimiento (Tabla 13). El ecocardiograma Doppler provee varios parámetros que correlacionan con el pronóstico de la enfermedad. El derrame perciárdico se asocia a peor perfil hemodinámico, su persistencia tras el tratamiento demostró ser un marcador independiente de disminución de supervivencia64. En ocasiones, cuando existe sospecha de comunicaciones interauriculares o interventriculares la realización de un ecocardiograma contrastado con burbujas permite detectar o descartar la patología. En la Tabla 14 se describen otros hallazgos ecocardiográficos que sugieren HP. Para acceder a una guia detallada de la evaluación ecocardiográfica del corazón derecho se remite al lector a la Guía de la European Association of Cardiovascular Imaging (EACVI).


El ecocardiograma tiene como ventajas ser un método no invasivo, reproducible, relativamente económico y fácil de obtener. Sus desventajas incluyen la imposibilidad de calcular la PPs en caso de ausencia de regurgitación tricuspídea y la baja correlación entre este valor estimado y el valor medido por CCD en algunas patologías. Asimismo, no permite calcular directamente la RVP ni la PAPE. En el caso de insuficiencia tricuspídea masiva y obstrucción tracto salida ventrículo derecho no existe correlación.
2.5.1 Ecocardiograma transesofágico
En algunas oportunidades es necesario recurrir al ecocardiograma transesofágico. Este estudio es útil para evaluar las aurículas y las válvulas auriculoventriculares, la integridad del septum interauricular y para detectar el pasaje de burbujas a través de un potencial defecto del septum. También se emplea para examinar las venas pulmonares, e investigar la presencia de trombos67, 68. El principal aporte de este estudio es descartar comunicación interauricular, anomalías del retorno venoso y valorar la cuantia de la insuficiencia mitral (Eco no diagnóstico)
2.6 Laboratorio de función pulmonar e intercambio gaseoso
La espirometría en la HAPI es en la mayoría de los casos normal; en otras ocasiones puede mostrar obstrucción leve de la vía aérea. Puede también hallarse leve a moderada reducción de los volúmenes pulmonares medidos mediante pletismografia (Tabla 15)10, 69, 70.

El hallazgo más frecuentemente identificado en la HP es una reducción de la capacidad de difusión con monóxido de carbono (DLCO), un valor < 45% es signo de mal pronóstico(10). Este hallazgo requiere aún más esfuerzo para poder discriminar cuánto de dicha reducción es producida por la vasculopatía pulmonar y cuánto se debe al agregado de un componente intersticial cuando se trate de HAP asociada a ES o HP secundaria enfermedades del parénquima pulmonar, EVOP o HCP (Tabla 15). Se destaca la relevancia de interpretar los resultados del laboratorio de función pulmonar en el contexto de las imágenes radiológicas10.
En aquellas enfermedades que producen HP asociadas con enfermedades pulmonares, los hallazgos del laboratorio de función pulmonar orientan a las patologías que la acompañan2-5, 10. En caso de sospecharse apnea obstructiva del sueño o hipoventilación se recomienda realizar oximetría nocturna, poligrafía respiratoria o polisomnografía10.
2.7 Pruebas de esfuerzo
2.7.1 Prueba de marcha de seis minutos (PM6M)
La PM6M es una prueba submáxima técnicamente simple, reproducible y bien estandarizada. Se reconoce una relación entre la distancia recorrida en la evaluación basal y la supervivencia, una distancia recorrida en la evaluación basal inferior a 332 mt se ha correlacionado con peor pronóstico. Resultados similares se obtuvieron en pacientes tratados con infusión de epoprostenol con una distancia recorrida inicial < 250 metros, en quienes se notó asociación con una menor tasa de supervivencia72. El incremento en los metros recorridos es considerado como un objetivo importante para evaluar el efecto del tratamiento. Las Guías de Niza y las ESC/ERS adoptan un umbral de 440 metros derivado de la mayor cohorte estudiada; sin embargo se recomienda adaptar las exigencias a cada paciente dado que en individuos de más edad o con comorbilidad pueden aceptarse distancias más cortas, en tanto que esta distancia podría parecer corta en jóvenes10. También se ha propuesto que este objetivo es difícil de lograr en pacientes que tienen enfermedad poco avanzada (“ceiling effect”, efecto cielo o techo) (estudio EARLY). Sobre esta base se resalta la importancia de una evaluación integral. A propósito, el metaanálisis de Savarese y cols (JACC, 2012), con 22 ensayos clinicos, refleja una escasa correlación entre el aumento de la distancia recorrida y endpoints clínicamente relevantes que reflejan morbilidad.
2.7.2 Prueba de ejercicio cardiopulmonar
Este estudio es el que mejor define la condición cardiorrespiratoria del paciente a través de la medición de los siguientes parámetros funcionales: consumo máximo de oxígeno (VO2máx), umbral anaeróbico (VO2AT), pulso de oxígeno, equivalente ventilatorio del dióxido de carbono (VE/VCO2), espacio muerto (VD/VT), presión end-tidal CO2 (PETCO2) (Tabla 16)73. No se recomienda la realización de este estudio en pacientes con antecedentes de síncope, con presión sistólica sistémica < 90 mmHg y clase funcional grado IV5. Se ha establecido que el VO2máx es un predictor pronóstico independiente, así como la presión sistólica sistémica al VO2máx. Estas pruebas están específicamente recomendadas en los más jóvenes ya que aportan información objetiva sobre la función del VD y la capacidad de ejercicio10.
El hallazgo más frecuentemente identificado en la HP es una reducción de la capacidad de difusión con monóxido de carbono (DLCO), un valor < 45% es signo de mal pronóstico(10). Este hallazgo requiere aún más esfuerzo para poder discriminar cuánto de dicha reducción es producida por la vasculopatía pulmonar y cuánto se debe al agregado de un componente intersticial cuando se trate de HAP asociada a ES o HP secundaria enfermedades del parénquima pulmonar, EVOP o HCP (Tabla 15). Se destaca la relevancia de interpretar los resultados del laboratorio de función pulmonar en el contexto de las imágenes radiológicas10.
En aquellas enfermedades que producen HP asociadas con enfermedades pulmonares, los hallazgos del laboratorio de función pulmonar orientan a las patologías que la acompañan2-5, 10. En caso de sospecharse apnea obstructiva del sueño o hipoventilación se recomienda realizar oximetría nocturna, poligrafía respiratoria o polisomnografía10.
2.7 Pruebas de esfuerzo
2.7.1 Prueba de marcha de seis minutos (PM6M)
La PM6M es una prueba submáxima técnicamente simple, reproducible y bien estandarizada. Se reconoce una relación entre la distancia recorrida en la evaluación basal y la supervivencia, una distancia recorrida en la evaluación basal inferior a 332 mt se ha correlacionado con peor pronóstico. Resultados similares se obtuvieron en pacientes tratados con infusión de epoprostenol con una distancia recorrida inicial < 250 metros, en quienes se notó asociación con una menor tasa de supervivencia72. El incremento en los metros recorridos es considerado como un objetivo importante para evaluar el efecto del tratamiento. Las Guías de Niza y las ESC/ERS adoptan un umbral de 440 metros derivado de la mayor cohorte estudiada; sin embargo se recomienda adaptar las exigencias a cada paciente dado que en individuos de más edad o con comorbilidad pueden aceptarse distancias más cortas, en tanto que esta distancia podría parecer corta en jóvenes10. También se ha propuesto que este objetivo es difícil de lograr en pacientes que tienen enfermedad poco avanzada (“ceiling effect”, efecto cielo o techo) (estudio EARLY). Sobre esta base se resalta la importancia de una evaluación integral. A propósito, el metaanálisis de Savarese y cols (JACC, 2012), con 22 ensayos clinicos, refleja una escasa correlación entre el aumento de la distancia recorrida y endpoints clínicamente relevantes que reflejan morbilidad.
2.7.2 Prueba de ejercicio cardiopulmonar
Este estudio es el que mejor define la condición cardiorrespiratoria del paciente a través de la medición de los siguientes parámetros funcionales: consumo máximo de oxígeno (VO2máx), umbral anaeróbico (VO2AT), pulso de oxígeno, equivalente ventilatorio del dióxido de carbono (VE/VCO2), espacio muerto (VD/VT), presión end-tidal CO2 (PETCO2) (Tabla 16)73. No se recomienda la realización de este estudio en pacientes con antecedentes de síncope, con presión sistólica sistémica < 90 mmHg y clase funcional grado IV5. Se ha establecido que el VO2máx es un predictor pronóstico independiente, así como la presión sistólica sistémica al VO2máx. Estas pruebas están específicamente recomendadas en los más jóvenes ya que aportan información objetiva sobre la función del VD y la capacidad de ejercicio10.

2.8 Tomografía computada y angiotomografía
2.8.1 Tomografía axial computada (TAC) y tomografía axial computada de alta resolución (TACAR)
Tanto la TAC como la TACAR orientan el diagnóstico en pacientes con sospecha de HP, y contribuyen a identificar enfermedades involucradas en la fisiopatogenia de la HP (Tabla 17) En particular la TACAR es muy útil en caso de sospecha clínica de EPVO y HCP (Tabla 18)5, 7, 10, 74.


2.8.2 Angiotomografía con contraste
Este estudio es útil para diagnosticar cortocircuitos arteriovenosos pulmonares, tromboembolismo pulmonar agudo y crónico recurrente ya que pone en evidencia las obstrucciones e irregularidades de los vasos pulmonares; y, asimismo, orienta acerca de la posibilidad de un acceso quirúrgico5, 10, 75, 76.
2.9 Medicina nuclear
2.9.1 Centellograma ventilación/perfusión (V/Q)
Debe realizarse siempre en la evaluación diagnóstica de la HP a fin de descartar la HPTEC10, 77. Un estudio normal o de baja probabilidad excluye HPTEC con una sensibilidad del 90-100% y una especificidad del 94% al 100%; sin embargo, muchos de estos exámenes no son suficientes para establecer el diagnóstico ya que algunos pequeños defectos pueden también estar presentes en la EPVO10, 78, 79. En la HAPI el estudio es habitualmente normal o de baja probabilidad con una imagen de piqueteado difuso “en sal y pimienta” en la perfusión5. No se utiliza el centellograma V/Q para el diagnóstico de las HAP asociadas a EVOP y HCP. Los criterios diagnósticos en este subgrupo incluyen la imagen de la TACAR que es sumamente característica, asociada a otros exámenes complementarios10.
2.10 Angiografía pulmonar
La angiografía pulmonar continúa siendo el procedimiento “de oro” para definir la anatomía vascular pulmonar y confirmar el diagnóstico de HPTEC. Permite visualizar defectos de llenado que traducen los trombos endoluminales y las alteraciones en la vascularización pulmonar. Se recomienda su utilización en pacientes con HPTEC para determinar la extensión y localización de los trombos lo cual contribuye a identificar a los potenciales beneficiarios de una endarterectomía pulmonar o angioplastia pulmonar con balón5, 10. En la HAPI las arterias pulmonares están dilatadas con un brusco afinamiento e hipovascularización periférica mostrando una imagen en “árbol de invierno”. Permite además el diagnóstico de malformaciones vasculares y vasculitis. La angiografía pulmonar no está contraindicada en los enfermos con HP severa y puede realizarse en forma segura en servicios con experiencia, mediante la inyección de contraste selectivo, tomando las medidas de seguridad apropiadas5, 10, 80. En el Simposio de Niza se estableció que este examen puede formar parte del CCD pero no debe realizarse hasta después de haberse completado la totalidad de las evaluaciones hemodinámicas9.
2.11 Resonancia magnética nuclear
La resonancia magnética nuclear (RMN) cardíaca es el gold standard para la evaluación de la función del ventrículo derecho. Es un método confiable para visualizar el tamaño, masa, forma y volumen del VD, el volumen sistólico y diastólico y el volumen de eyección del VD, además de la distensibilidad de la arteria pulmonar. Permite la valoración dinámica de la arteria pulmonar principal relacionada con respuesta vasodilatadora aguda81. También es útil para evaluar el colapso de las cavidades izquierdas por el movimiento paradojal del septum interventricular. Asimismo, puede detectar cardiopatías asociadas con HP como enfermedades infiltrativas del VI, miocardiopatía hipertrófica y cardiopatías congénitas, y drenajes venosos anómalos5, 82. En pacientes con HAP aporta información con valor pronóstico10. La angio-RMN puede ser utilizada en mujeres embarazadas, y pacientes con alergia al contraste iodado con sospecha de tromboembolismo83.
2.12 Ultrasonografía
2.12.1 Ecografía Doppler abdominal
En presencia de hepatopatías, especialmente cirrosis con hipertensión portal, la ecografía Doppler abdominal está indicada para diagnosticar hipertensión portal que pueda producir HP asociada a la misma, aunque un resultado normal no excluye el diagnóstico10. Este examen permite detectar cortocircuitos portosistémicos5, 84.
2.12.2 Fibroscan o elastografía hepática
Esta técnica ultrasonográfica se basa en la elastografía, mide la velocidad de propagación de ondas elásticas a través del hígado y sirve para evaluar el grado de fibrosis hepática. La elastografía valora el estado de rigidez o elasticidad de un tejido utilizando una onda de ultrasonido (5MHz) y un pulso mecánico de vibración de baja frecuencia (50 Hz). Los resultados obtenidos se expresan en kilopascal (kPa). Valores > 7kPa indican la existencia de fibrosis85.
2.13 Cateterismo cardíaco derecho
El CCD permite confirmar el diagnóstico de HP, distinguir si la HP es precapilar o poscapilar, evaluar su gravedad hemodinámica y realizar, cuando esté indicada, la prueba de vasorreactividad pulmonar aguda (PVRPA) y la angiografía pulmonar convencional. En el seguimiento, permite analizar la eficacia del tratamiento y, de ser necesario, optimizarlo de manera objetiva intensificándolo o combinándolo con otras drogas. También es útil para detectar un eventual deterioro clínico (Tablas 19 y 20) Realizado en centros de referencia, el procedimiento tiene morbilidad y mortalidad bajas y un rédito muy alto5, 9, 10, 86.
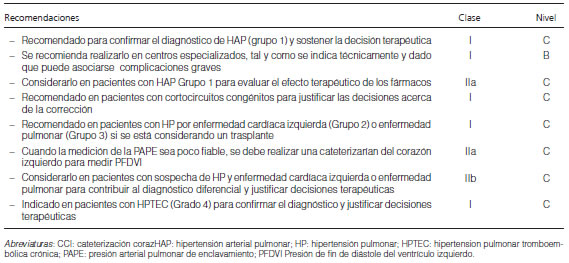
La saturación de oxigeno (SO2) debe medirse en sangre de las venas cavas superior e inferior, la arteria pulmonar y la sangre sistémica arterial. En pacientes con SO2 >75% en la arteria pulmonar o sospecha un cortocircuito de izquierda a derecha se recomienda una evaluación escalonada.
La resistencia vascular pulmonar (RVP) se calcula tomando la diferencia entre la PAPm y la presión de oclusión de la arteria pulmonar (POAP) o PAP capilar pulmonar, dividiéndola por el VM (VM) cardíaco. Se recomienda la utilización de UW (mmHg/litros/minuto) y no utilizar UI. El límite superior normal de la RVP es de 2 WU; no obstante el valor de corte para la HAP es de 3 UW dado que pacientes con valores inferiores tienen escasas probabilidades de padecer HAP5, 9, 87.
El GDTP es la diferencia entre la PAP diastólica y la PAP capilar, su valor normal es < 7 mmHg.
El índice cardíaco (IC) se obtiene dividiendo el VM por la superficie corporal y se expresa en litros/ minuto/m2.
La resistencia vascular sistémica (RVS) es la diferencia entre la presión arterial media y la presión de la aurícula derecha dividida por el VM.
El VM se mide con el método de termodilución (estándar de oro) que provee mediciones confiables aún en pacientes con VM muy bajo o regurgitación tricuspídea severa (Tabla 20)10, 88. En pacientes con comunicaciones interauriculares o ventriculares se recomienda determinar el VM por el método de Fick. En pacientes con cortocircuito intracardíaco, la termodilución puede ser inexacta debido a la recirculación temprana del inyectado. El método de Fick directo requiere la medición directa del consumo de O2, una técnica que no está ampliamente disponible. El método de Fick indirecto, que utiliza los valores estimados de consumo de O2, es aceptable, pero carece de fiabilidad.
2.13.1 Prueba de sobrecarga con fluidos
Esta prueba requiere una meticulosa evaluación y estandarización antes de ser recomendada rutinariamente en la práctica clínica9, 10. En pacientes con enfermedad cardíaca izquierda el resultado del CCD puede ser erróneo, debido a que la PAPE se puede reducir a < 15 mmHg con diuréticos10. Por ello es que debe considerarse una prueba de sobrecarga con fluidos. Una de las modalidades consiste en la administración de 500 ml de solución salina en un período de 5 a 10 minutos y puede ser útil para establecer el diagnóstico diferencial entre HAP y HP por disfunción diastólica del VI, al incrementar la PAPE en esta última. Por el momento, no hay un protocolo recomendado.
2.13.2 Prueba de provocación por ejercicio
Es posible que la hemodinamia durante el ejercicio pueda ser útil para poner de manifiesto una disfunción del VI. Sin embargo, se necesitan nuevas evaluaciones, estandarización y comparación con la prueba de sobrecarga de volumen antes de recomendar su uso en la práctica clínica9, 10, 89-91.
2.14 Prueba de vasorreactividad pulmonar aguda
La PVRPA está indicada para la caracterización hemodinámica de pacientes con HAPI, HAPH y HAPD con el objetivo de identificar potenciales beneficiarios del tratamiento con altas dosis de bloqueantes de los canales del calcio (BCC) (Tabla 21)5, 9, 10.

Consideraciones:
– Suspender el tratamiento de los fármacos especificos para HAP con una antelación de 2,5 vidas medias antes de realizar la prueba.
– Contraindicada en pacientes con disfunción severa de VD, ya que per se tienen contraindicación de recibir BCC, y además, se incremetan las complicaciones vinculadas a la alteración abrupta del VM, más probable en este grupo de pacientes.
– Existen varios agentes para realizar esta prueba, lo ideal es que sea selectivo para la circulación pulmonar con un comienzo y finalización de acción rápidos (Tablas 21 y 22). El óxido nítrico (ON) es el estándar de oro para la PVRPA, por ser mejor tolerado y requerir menos tiempo5, 9, 10, 87.
– No se recomienda el uso de BCC en esta prueba, tampoco IPDE-5 ni O2.
– Unicamente el 10% de los pacientes con HAPI tiene una respuesta positiva (Tabla 21)5, 9, 10.
– Las Guías Europeas ESC/ERS 2015 consideran que los pacientes con HAPI con criterios de una respuesta vasodilatadora positiva tratados con BBC deberían ser monitoreados estrechamente por razones de seguridad y eficacia con reevaluaciones completas después de tres a cuatro meses de tratamiento, incluyendo el CCD.
– Un 50% de los respondedores agudos serán también respondedores a largo plazo y podrán mantenerse en el tiempo con BCC como monodroga; para el resto, será necesario modificar el tratamiento27.

2.15 Cateterismo cardíaco izquierdo
El cateterismo cardíaco izquierdo (CCI) no está indicado en la práctica de rutina y, las pruebas hemodinámicas invasivas deben contextualizarse en función de la clínica y los hallazgos ecocardiográficos9. El umbral para su indicación debería ser bajo en pacientes con factores de riesgo de enfermedad coronaria, insuficiencia cardíaca con fracción de eyección conservada y/o signos de disfunción diastólica del VI en el ecocardiograma. Se considera importante medir la presión de fin de diástole del VI para evitar errores de clasificación en pacientes con PAPE inesperadamente alta sin compromiso aparente de la función cardíaca izquierda10.
2.16 Cinecoronariografia
La cinecoronariografia no representa una indicación convencional, excepto en pacientes con angina de pecho, factores de riesgo de enfermedad coronaria, disfunción ventricular izquierda sistólica significativa, o en lista de espera para trasplante pulmonar y candidatos a endarterectomía pulmonar10.
2.17 Pruebas genéticas
El acceso a las pruebas genéticas y el consejo genético plantea un dilema ético que, por el momento, puede subsanarse remitiéndose a las disposiciones legales vigentes en cada país (Tabla 23)10, 92.

2.18 Biopsia pulmonar
La biopsia pulmonar quirúrgica toracoscópica o a cielo abierto se asocia a una elevada tasa de morbimortalidad y, es poco probable que modifique el diagnóstico y tratamiento. No está recomendada en pacientes con HAP10. Sin embargo, puede considerarse frente a la sospecha de microangiopatía pulmonar, HCP o EPVO si la condición del paciente lo permite. Asimismo, puede demostrar la presencia de vasculitis, enfermedad granulomatosa o enfermedad pulmonar intersticial5, 93, 94. Sólo se recomienda en aquellos casos en que no haya sido posible confirmar vasculitis o enfermedad granulomatosa por otros medios (imágenes o laboratorio).
2.19 Algoritmo diagnóstico
En el Simposio de Niza se propuso un algoritmo diagnóstico el cual ha sufrido algunas modificaciones en el último consenso ESC/ERS del año 2015. Uno de los puntos más destacados ha sido la importancia de derivar a los pacientes a centros de referencia especializados en el manejo de la HP, tanto para el diagnóstico como para el tratamiento (Tabla 24 y Figura 1)10.

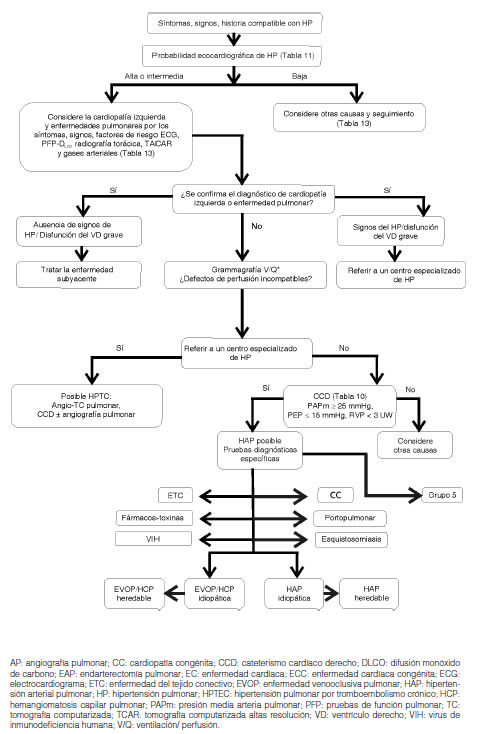
Consideraciones
– El CCD es mandatario para confirmar el diagnóstico de HP.
– Si la PAPm es ≥ 25mmHg, la PAPE es ≤ 15 mmHg y la RVP es > 3UW se tratará de una probable HAP, debiéndose realizar pruebas específicas de diagnóstico a fin de definir la etiología y la clasificación de la HP.
– El diagnóstico de HAP requiere la exclusión de otras causas de HP.
– Para el diagnóstico de ETC, además de las manifestaciones clínicas y los criterios diagnósticos, deben solicitarse los anticuerpos específicos.
– El diagnóstico de HAPD se basa en el antecedente de exposición.
– Para la HAP asociada a infección por VIH, además de los antecedentes, son fundamentales la serología para virus VIH en sus distintas modalidades (ELISA, Western Blot y antigenemia P24).
– Para el diagnóstico de HP asociada a patología portopulmonar son necesarias las pruebas de función hepática, la ecografía hepatoesplénica con ecografía Doppler abdominal, y CCD con medición de presión de las venas suprahepáticas y gradiente de presión portal.
– El diagnóstico de esquistosomiasis debe ser considerado en las áreas endémicas, y requiere una clínica compatible, determinación de anticuerpos y eventual biopsia hepática.
– Si todos estos pasos han dado negativos estaremos en presencia de HAPI o HAPH, en cuyo caso, en base a los antecedentes familiares se deberá considerar la realización de estudios genéticos en centros de referencia9.
– Las patologías asociadas a HP incluidas en el Grupo 5 deben ser descartadas minuciosamente.
3. Hipertensión arterial pulmonar grupo 1
3.1 Evaluación y Pronóstico
Si bien la signosintomatología es semejante para todas las entidades que causan HAP, el pronóstico está significativamente determinado por su etiología. Las asociadas a cardiopatías congénitas tienen el mejor pronóstico; le sigue la HAPI y, en menor medida, la secundaria a ETC. La asociada a EPVO es la de peor evolución5, 95, 96. Desde la ecuación de pronóstico realizada por D’Alonzo en 1991, se han publicado otras varias97, 98, 99, validadas en reiterados estudios. En las Guías ESC/ERS 2015 se seleccionan algunas variables a partir de las cuales se definen categorías de riesgo aclarándose que los valores de corte se establecieron en base a opiniones de expertos y que la mayoría de ellos se validaron en pacientes con HAPI10.
3.1.1 Parámetros clínicos
En cada visita es indispensable evaluar:
– cambios en la capacidad de ejercicio
– dolor torácico
– alteraciones del ritmo cardíaco
– hemoptisis
– síncope
– cianosis
– ingurgitación yugular
– edema
– ascitis
– derrame pleural
– presión arterial
– adherencia con las indicaciones
– comorbilidad
– complicaciones
– eventuales cambios en la medicación
3.1.2 Capacidad funcional
La evaluación de la clase funcional (CF) New York Heart Association (NYHA) o de la Organización Mundial de la Salud (OMS) es una forma sencilla de evaluar la gravedad y se correlaciona con el pronóstico (Tabla 25). Proporciona información sobre la tolerancia al ejercicio y la calidad de vida. En el registro del National Institutes of Health, se encontró que la CF estaba íntimamente relacionada con la supervivencia, cuya mediana a los seis meses en los pacientes en CF I-II y III- IV era de 6 y 2,5 años, respectivamente5, 96. Otros estudios demostraron que la CF basal también tenía impacto pronóstico en pacientes con HAPI tratados posteriormente con prostanoides endovenosos, evidenciándose una supervivencia a los 3 y 5 años, mayor en los pacientes en CF III que en aquellos en CF IV100. Asimismo, la persistencia en CF III-IV después de tres meses de tratamiento con epoprostenol se relacionó con elevada mortalidad a 3 y 5 años5, 7.

3.1.3 Ecocardiograma
La presión arterial pulmonar sistólica (PAPS) estimada a partir de la velocidad de regurgitacion tricuspídea en reposo no refleja progresión ni mejoría, carece de valor pronóstico y de relevancia para las decisiones terapéuticas. Por el contrario el ecocardiograma es fundamental para el diagnóstico y seguimiento. En relación a los parámetros ecocardiográficos, el derrame pericárdico se ha relacionado consistentemente con el pronóstico en los pacientes con HAPI. Su magnitud se correlaciona con muerte o necesidad de trasplante pulmonar al año5, 101. El área de la aurícula derecha (AD) también es predictora de supervivencia. En los últimos años, se ha incorporado como parámetro pronóstico la excursión sistólica del anillo tricuspídeo (TAPSE sigla en inglés), valores <18 mm se correlacionan con mayor disfunción del VD y, por ende, con peor pronóstico. Pacientes con un TAPSE <18 mm presentan una supervivencia a 1 y 2 años, de 60% y 50%, respectivamente, comparada con 94% y 88% en pacientes con TAPSE >18 mm. La determinación de la capacitancia vascular pulmonar por ecocardiografía parece agregar información a los parámetros tradicionales5, 102. El ecocardiograma durante el ejercicio puede brindar información adicional, un aumento de la PAP > 30 mmHg refleja la función del VD y se asocia con buena evolución a largo plazo (reserva contráctil)10.
3.1.4 Resonancia magnética cardiaca
La RMN cardiaca es altamente confiable para evaluar forma y función del VD, tiene valor pronóstico y contribuye a las decisiones terapéuticas. Se consideran marcadores de mal pronóstico: aumento del volumen VD, disminucion volumen VI, disminucion fracción de eyección VD, reducción volumen de choque VD10. La RNM cardiaca es el gold standard para la evaluación de la función del VD aunque por su limitada disponibilidad, su uso es poco difundido. Sin embargo es de gran utilidad en la evaluación de un paciente que fracasa a terapias y que se postula para trasplante.
3.1.5 Parámetros hemodinámicos
El CCD tiene valor pronóstico. Las variables que ofician de indicadores de función del VD y pronóstico son la presión de la AD, el IC y la SvO210. El incremento de la presión de la AD y la caída del IC se asocian a mayor mortalidad. El aumento de la PAP tiene un valor pronóstico más débil en relación a la supervivencia26. Existen controversias acerca del momento en que debe realizarse el CCD y del valor de este método invasivo para el seguimiento; no obstante, hay acuerdo acerca de la necesidad de realizarlo cuando sus resultados pudieran determinar cambios en el tratamiento10, 103.
3.1.6 Capacidad de ejercicio
3.1.6.1 Prueba de la distancia caminada en seis minutos
La PM6M es sencilla, económica y con un fuerte valor pronóstico, si bien debe ser interpretada en el contexto clínico dado que sus resultados son influenciables por numerosas variables (edad, sexo, etc). Muestra muy buena correlación con la clase funcional en todas las formas de HP12. La distancia caminada es menor en los pacientes que fallecen a corto plazo que en aquellos que sobreviven, habiéndose señalado valores de corte de 332 metros71 o 380 metros72, siendo la distancia actual para discriminar pronóstico de 440 metros. En su metanálisis de 22 estudios Savarese y cols (2012) señala la inexistencia de una adecuada correlación entre el aumento de la distancia recorrida bajo terapia y variables clínicas de morbimortalidad y propone no adoptar esta prueba como endpoint sustituto ni único marcador de evolución.
3.1.6.2 Prueba de ejercicio cardiopulmonar
Aporta información importante sobre la capacidad de ejercicio y el intercambio de gases, la eficacia de la ventilación y la función cardiaca durante el ejercicio. La prueba no está estandarizada (Tabla 16)10. Un consumo máximo de O2 menor de 15 ml/kg/min o un valor menor del 65% del predicho o una presión arterial sistólica máxima < 120 mmHg durante la actividad son predictores independientes de peor evolución5, 104, aunque estos resultados están pendientes de confirmación10.
3.1.7 Biomarcadores
El BNP y su fragmento biológicamente inactivo el NT-proBNP son los únicos usados en la práctica clínica de rutina. Secretados principalmente por los ventrículos, su aumento indica disfunción ventricular derecha e izquierda. Valores elevados de NT-proBNP se asociaron a menor capacidad de ejercicio, peor clase funcional y a disfunción del VD; un valor > 1400 pg/ml indica peor pronóstico105. Concentraciones de BNP >150 pg/ml, así como su incremento durante el seguimiento (>180 pg/ml) se relacionaron con mayor mortalidad106. Contrariamente, un descenso > 50% durante los primeros tres meses de tratamiento con epoprostenol es un potente predictor de supervivencia libre de eventos107. Los valores de BNP y de NT-proBNP pueden estar influenciados por otras condiciones como insuficiencia renal, enfermedad coronaria e insuficiencia cardíaca izquierda, por lo que deben ser interpretados cuidadosamente. El NT-proBNP ofrece como ventaja una estabilidad y una exactitud interna mayores que el BNP.
3.1.8 Otras evaluaciones
– ECG para detectar arritmias
– Gases en sangre: la hipoxemia es frecuente, se asocia con disminucion del flujo pulmonar, sugiere oxigenoterapia y es indicador de mal pronóstico. También la hipocapnia es relevante en su valor pronóstico.
– RIN (INR) en pacientes tratados con antagonistas de la vitamina K
– Electrolitos, creatinina y ácido úrico
– AST (amino aspartato transferasa) también llamada GOT (glutamato-oxalacetato transaminasa), y ALT (alanina amino transferasa) también llamada GPT (glutamato piruvato transaminasa) en pacientes tratados con antagonistas de receptores de endotelina
– Troponina, acido úrico, ferremia, función tiroidea anualmente o en caso de peoría.
3.1.9 Consideraciones
– La evaluación sirve para establecer la gravedad de la enfermedad y orientar la elección del tratamiento; el seguimiento es importante para definir la respuesta terapéutica y estimar el pronóstico9.
– Se recomienda evaluar periódicamente la gravedad de los pacientes con HAP con un conjunto de estudios clínicos, bioquímicos, imagenológicos, pruebas de ejercicio, ecocardiograma y pruebas hemodinámicas (Nivel de recomendación I- Clase C) (ver Tabla 26)10.
– Estas evaluaciones se llevarán a cabo cada tres a seis meses en los pacientes estables (Nivel de recomendación I- Clase C) (ver Tabla 26)10.
– Se definieron criterios de estabilidad clínica con utilidad para determinar respuesta al tratamiento especulando con que aquellos que alcancen estos objetivos tendrán un mejor pronóstico (ver Tabla 27)9, 10. En las Guías ESC/ERS 2015 estos criterios han sido ampliados estableciéndose categorías de riesgo de mortalidad. Al respecto se establece que alcanzar y mantener un perfil de bajo riesgo define una respuesta terapéutica adecuada (Nivel de recomendación I- Clase C) (Tabla 28)10.
– Entre los criterios de buen pronóstico se han modificado la distancia en la PM6M de 500 metros a más de 440 metros y se ha incluido el TAPSE >20 mm5, 10.
– En la mayoría de los pacientes con HAP, alcanzar y mantener un perfil de riesgo intermedio podría considerarse una respuesta terapéutica inadecuada (Nivel de recomendación IIa- Clase C) (Tabla 28)10.
– Se definieron indicadores de mal pronóstico o alto riesgo (Tabla 28)10.
– Se ha propuesto predecir la supervivencia de los pacientes con HAP a través de un score del Registro REVEAL que puede ayudar a la toma de decisiones relacionada con el manejo de esta afección. Este score ha sido útil para predecir la supervivencia al año. Este calculador evalúa datos demográficos y comorbilidades, clase funcional, presión arterial y frecuencia cardiaca, distancia caminada en la PM6M, valor del BNP, derrame pericárdico ecocardiográfico, valores de DLCO, y hallazgos del CCD. Acorde a su puntuación los pacientes se clasifican en bajo riesgo (score 1-7), con una supervivencia al año del 95-100%; riesgo promedio (score 8) 90 a < 95%, riesgo moderadamente alto (score 9) < 90% y hasta 85%, riesgo alto (score 10-11) < 85% y hasta 70%; riesgo muy alto (score 12 o más) < 70%108.


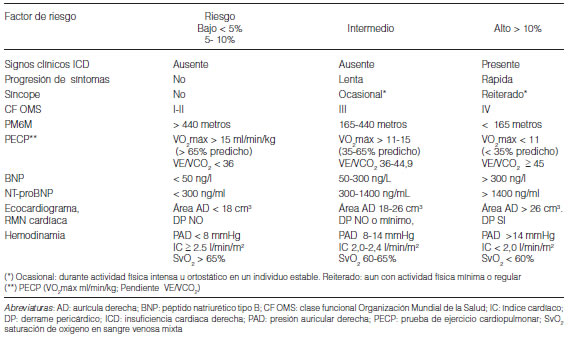
3.2 Medidas terapéuticas generales
La necesidad de definir los objetivos terapéuticos capaces de modificar el pronóstico a largo plazo de esta enfermedad fue uno de los temas clave en el Consenso de Niza. Al respecto, se han enumerado una serie de parámetros que, de ser alcanzados, independientemente del tratamiento recibido, se asocian a un mejor pronóstico (Ver arriba Tabla 27). El abordaje terapéutico de la HAP es complejo y se lo divide en tres etapas (Tabla 29).

3.2.1 Actividad física y rehabilitación supervisada
La HP restringe la capacidad física, afecta la calidad de vida y genera depresión y ansiedad. El entrenamiento se asocia a un incremento de la actividad física, aumento de la distancia caminada en seis minutos, función cardiorrespiratoria y mejoría de la calidad de vida, en comparación con aquellos no entrenados109-111. Los resultados han sido buenos pero los estudios han incluido pocos pacientes, se han desarrollado bajo una estricta supervisión y hasta en ámbitos de internación, lo que los aleja de la práctica clínica habitual. Las Guías 2015 ESC/ERS establecen un Nivel de recomendación IIa, Clase B para “la consideración de entrenamiento supervisado en pacientes con desacondicionamiento físico que reciben tratamiento médico”1. Debido a la gravedad de esta enfermedad, se recomienda iniciar el entrenamiento en el ámbito hospitalario o en un centro de rehabilitación especializado en enfermedades cardiovasculares y respiratorias graves. La actividad debería ser suave y progresiva, con baja carga y una frecuencia de 4-5 días a la semana en pacientes con estabilidad clínica y bajo el óptimo tratamiento farmacológico recomendado2, 5, 109, 112-114. Se desaconseja realizar actividades físicas con riesgo de síntomas graves (síncope, lipotimia) o desaturación (III-C)10.
3.2.2 Embarazo, control de la natalidad y terapia hormonal sustitutiva en la posmenopausia
Se recomienda a las pacientes con HAP evitar el embarazo, la Joint Tak Force considera a esta recomendación (Clase I y le adjudica un Nivel de evidencia C)10. No obstante, en caso de producirse el embarazo podrían tratarse con terapias especificas para HAP y, llegado el momento del nacimiento realizar una cesárea planificada. Un estudio clínico reciente indica buena evolución en estas pacientes en particular en grupo de respondedoras de BCC115. En lo relacionado con los métodos anticonceptivos los de barrera son los más seguros para la paciente, pero pueden fallar por lo que se sugiere doble mecanismo de barrera y hormonal (preservativo, anillo vaginal, dispositivo intrauterino con progestágenos, progrestágenos inyectables). Un dato importante es que el bosentán disminuye la eficacia de los anticonceptivos orales; en tanto el dispositivo intrauterino con levonorgestrel puede causar reacción vasovagal en el momento de su inserción10. Acerca de la terapia hormonal sustitutiva, se reserva su indicación a los casos con síntomas mal tolerados y en asociación con anticoagulación por vía oral10.
3.2.3 Cirugía electiva
Los pacientes con HP tendrían mayor riesgo de complicaciones intraoperatorias y postoperatorias. A la fecha, no existen datos disponibles para determinar la mejor estrategia anestésica, pero se admite que, probablemente, la anestesia epidural sea mejor tolerada que la anestesia general (IIa-C)10. Los pacientes deben pasar paulatinamente del tratamiento oral al intravenoso o nebulizado hasta que sean capaces de deglutir y absorber las drogas administradas por vía oral10, 116.
3.2.4 Prevención de las infecciones
La neumonía es una de las causas de mortalidad en pacientes con HAP. Se recomienda la inmunización contra influenza y neumococo (I-C)10. También se recomienda la vacunación contra la hepatitis7, 117.
3.2.5 Tratamiento de la anemia
La deficiencia de hierro es un hallazgo frecuente en pacientes con HAP,-. Se recomienda monitorear periódicamente este parámetro y considerar la prescripcion de hierro debido a que compromete la capacidad funcional de los pacientes (IIb-C)10.
3.2.6 Soporte psicosocial
Muchos pacientes con HP desarrollan ansiedad y depresión, además de trastornos de la memoria y la atención con el consiguiente deterioro en su calidad de vida5, 118, 119. Los grupos de ayuda de pacientes con HP podrían jugar un rol importante y debería estimularse a los pacientes a este fin (I-C)10.
3.2.7 Viajes en avión y zonas de altura
Durante desplazamientos aéreos o permanencia en regiones de altura se recomienda la provision de O2 suplementario en vuelo y zonas de altura a fin de disminuir la vasoconstricción hipóxica (Ver Oxigenoterapia).
3.3. Tratamiento farmacológico inespecífico
3.3.1 Anticoagulantes orales
Existen evidencias de lesiones trombóticas en HAPI, así como alteraciones de la coagulación y la fibrinólisis y, se han comunicado resultados favorable sobre el uso de anticoagulantes orales en la HAPI, HAPH y HAP asociada a anorexígenos120, 121, en los que se observó una mejoría en la supervivencia comparada con aquellos pacientes no tratados. Para los pacientes con HAPI se recomienda un RIN 1,5 a 2,55. Las Guías ESC/ERS 2015 recomiendan prescribir anticoagulantes en pacientes HAPI, HAPH y HAP asociada a anorexígenos (IIb-C)10. La evidencia no es fuerte porque la información viene de registros y no de ensayos randomizados. Los registros europeos COMPERA y el Norteamericano REVEAL sólo mostraron beneficio en el grupo idiopático y deterioro en los pacientes con esclerodermia en los que no se deben usar. La experiencia está restringida a la utilización de anticoagulantes orales antivitamina K. No existe experiencia con los nuevos anticoagulantes orales directos (DOACs).
3.3.2 Digoxina
Si bien es escasa la evidencia en favor del uso crónico de digoxina en la HP, se ha observado un aumento agudo del VM en pacientes con HAPI. Se considera que podría ser beneficiosa en individuos con HAPH y taquiarritmias auriculares. Aunque las Guías ESC/ERS 2015 no emiten opinión al respecto10, algunos autores consideran su uso en pacientes portadores de arritmia supraventricular1, 5.
3.3.3 Diuréticos
Los diuréticos están indicados en pacientes con HAP con signos de insuficiencia cardiaca derecha y retención de fluidos (I-C)10. Contribuyen a mantener una precarga ventricular derecha óptima y aliviar los síntomas2-5, 10, 122. Es deseable el monitoreo de la función renal y el potasio. El tratamiento con antagonistas de la aldosterona requiere evaluación sistemática de los niveles plasmáticos de electrolitos. Su uso en la esclerodermia no está recomendado por la posibilidad de desencadenar crisis renal5.
3.3.4 Oxigenoterapia
La hipoxemia es leve en la mayoría de los pacientes con HAP, a excepción de aquellos con cardiopatias congénitas, foramen oval permeable y cortocircuito pulmonar-sistémico10. En todo caso, se atribuye la hipoxemia en la HAP a la disminución del VM, la desaturación en sangre venosa mixta, y la alteración del cociente V/Q5.
La administración de O2 puro en agudo es capaz de disminuir la RVP y la PAP5, 123,124 pero no hay ensayos clínicos aleatorizados que aporten evidencias del beneficio de la oxigenoterapia crónica domiciliaria a largo plazo. Sin perjuicio de ello y ante esta falta de evidencia, podría plantearse el uso de oxigenoterapia de la misma forma que en el EPOC, es decir cuando la SO2 fuese < 91%5, 10, 125. Al respecto, las Guías ESC/ERS 2015 se pronuncian a favor de la oxigenoterapia a largo plazo en pacientes con valores persistentes de PaO2 < 8 kPa (60 mmHg) (I-C)10. La oxigenoterapia ambulatoria se recomienda cuando hubiera beneficio sintomático y se corrigiera la desaturación durante el ejercicio5, 10, 125.
Se recomienda considerar el suministro de O2 durante los vuelos aéreos en pacientes CF OMS III-IV y, en aquellos con PO2 < 8 kPa (60 mmHg) (IIa- C)10, 126. También se aconseja esta conducta cuando suban a altitudes >1500-2000 m. Un flujo de oxígeno promedio de 2 l/min lleva los valores inspirados de oxígeno a los obtenidos al del nivel del mar.
3.3.5 Otros
Las Guías ESC/ERS 2015 no recomiendan la prescripción de inhibidores de la enzima convertidora de angiotensina, antagonistas de los receptor II de la angiotensina, beta bloqueantes e ibravadina, a excepción de que su uso se justifique por una condición comórbida (hipertensión arterial, enfermedad de las arterias coronarias, insuficiencia cardiaca izquierda) (III-C)10. Se advierte que la combinación de medicación específica para la HAP con antihipertensivos conlleva riesgo de hipotensión sistémica10.
3.4 Terapia farmacológica específica
3.4.1 Bloqueantes de los canales del calcio
Un tratamiento con altas dosis BCC está indicado, únicamente, en pacientes con HAPI, HAPH y HAPD con respuesta positiva en la PVRPA (I-C)10. En pacientes con HAP-VIH, HAP portopulmonar, HAP-ETC y EVPO un resultado positivo en la PVRPA no predice una respuesta favorable a los BCC, a largo plazo. La elección del BCC se basa, fundamentalmente, en la CF; en general se inicia con dosis bajas fraccionadas en varias tomas titulando lentamente hasta el máximo tolerado. En aquellos con bradicardia, la nifedipina (dosis de 120-240 mg/día y amlodipina (dosis de 20 mg/día) son los más apropiados. El diltiazem es de elección en pacientes con taquicardia (dosis de 240-720 mg/día). Los efectos limitantes de dosis son hipotensión y edema de miembros inferiores. Estos pacientes requieren monitoreo 3 a 4 meses más tarde, incluyendo un CCD (I-C)10. La continuidad del tratamiento con BCC está dada en aquellos pacientes que han logrado estar en CF OMS I-II con marcada mejoría, casi normalización, de los parámetros hemodinámicos (I-C)10. En pacientes CF OMS III-IV, sin mejoría hemodinámica se planteará la posibilidad de iniciar terapia con medicamentos específicos: antagonistas receptores endotelina (ARE), IPDE-5, estimulantes guadenilato ciclasa o prostanoides (análogos de la prostaciclina) (I-C)10. Los BCC no están indicados en pacientes sin respuesta positiva a la PVRPA o con resultados negativos, si bien se los suele usar en dosis estándar para tratar otras patologías tales como el Sindrome de Raynaud (III-C)10.
3.4.2 Antagonistas de los receptores de la endotelina
Ya sea causa o consecuencia, existen evidencias de la activación del sistema endotelina en plasma y pulmón de pacientes con HAP evidenciable a través de un incremento de la endotelina-1 (ET-1)127. La unión de la ET-1 a sus receptores A y B ubicados en la membrana celular del músculo liso de la vasculatura pulmonar determina vasoconstricción y mitogénesis. En términos generales, el efecto adverso más frecuente de los ARE es el aumento de las enzimas hepáticas por lo que se recomienda el monitoreo mensual.
Ambrisentán. Es un ARE con selectividad para el receptor A. Ensayos clínicos en pacientes con HAPI, HAP-ETC y HAP-VIH han mostrado alivio sintomático, mejoría de la capacidad de ejercicio, parámetros hemodinámicos y aumento del intervalo de tiempo hasta el deterioro clínico128. Entre sus efectos adversos se mencionan el aumento de enzimas hepáticas, de menor prevalencia con respecto al bosentán, y edema periférico.
Bosentán. Inhibe ambos receptores A y B de la ET-1 (antagonista dual). Varios ensayos clínicos (BREATHE-2, BREATHE-5, EARLY y COMPASS-2) en pacientes con HAPI, HAP-ETC, HAP-Eisenmenger muestran mejoría de la capacidad de ejercicio, CF, parámetros hemodinámicos, ecocardiográficos y aumento del intervalo de tiempo hasta el deterioro clínico129-131. Bosentán es un inductor de las enzimas CYP3A4 y CYP2C9, por ende, la concentración plasmática de otros fármacos metabolizados por estas enzimas puede disminuir. Asimismo, puede aumentar la concentración plasmática de bosentán si se inhiben dichas enzimas por lo que se contraindica su coadministración con ketoconazol, ritonavir; amiodarona y fluconazol10. Es el más hepatotóxico, el efecto es dosis dependiente, con una incidencia del 10%. Por eso se recomienda el control mensual de la función hepática.
Macitentán. Inhibe ambos receptores ET-1 A y B (antagonista dual). En pacientes con HAP el uso de macitentán se asoció a una disminucion significativa de la morbilidad combinada (necesidad de septostomía atrial, trasplante, uso de prostanoides sistémicos, o deterioro clínico) y la mortalidad, con aumento significativo de la capacidad de ejercicio en comparación con placebo132. El efecto adverso más notable fue la anemia (descenso de la Hb < 8 gr/dl en un 4,3% de los pacientes).
3.4.3 Inhibidores de la fosfodiesterasa-5
La enzima fosfodiesterasa-5 se expresa abundantemente en los vasos pulmonares, su inhibición resulta en vasodilatación del árbol vascular pulmonar; además poseen efectos antiproliferativos y antiagregante plaquetario. Los efectos adversos de los IPDE-5 son leves a moderados y relacionados, fundamentalmente, con la vasodilatación (cefalea, epistaxis).
Sildenafil. Es un IPDE-5 potente y selectivo. En pacientes con HAP su uso se asocia con mejoría de la capacidad de ejercicio, los parámetros hemodinámicos y alivio sintomático10. La dosis aprobada es de 20 mg tres veces al día. Sildenafil es metabolizado por las enzimas CYP3A4 y CYP2C9; su biodisponibilidad aumenta si se coadministra con beta bloqueantes. Sus niveles séricos disminuyen al administrarlo conjuntamente con carbamazepina, fenitoína, fenobarbital, rifampicina e hipérico10.
Taladafil. Es un IPDE-5 selectivo. La dosis aprobada es 40 mg/día. En ensayos clínicos comparativos contra placebo, en pacientes con HAP, ha mostrado beneficios similares a los observados con sildenafil133, con la ventaja de ser utilizados en una única dosis. Se recomienda iniciar con una dosis de 20 mg titulando hasta 40 mg/día.
Vardenafil. Es un IPDE-5. Se administra en dos tomas diarias. En pacientes con HAP, vírgenes de tratamiento, ha mostrado un perfil de eficacia comparable a la de los anteriores. Este medicamento está pendiente de aprobación134.
3.4.4 Estimulantes de la guadenilato ciclasa.
Riociguat. Aumenta la producción de GMPc estimulando a la guadenilato ciclasa directamente, aún en ausencia del ON, y potenciando la acción de éste sobre dicha enzima; en modelos animales se han observado efectos antiproliferativos y antirremodelamiento. El esquema de titulación se inicia con tres dosis diarias de 1 mg con ajustes cada dos semanas, hasta alcanzar tres dosis diarias de 2.5 mg al final de una fase de ajuste de 8 semanas135. En el ensayo clínico PATENT en pacientes con HAP, tratados con prostanoides o ARE, se observó que, un esquema combinado con hasta tres dosis diarias de 2.5 mg de riociguat se asoció con mejoría de la capacidad de ejercicio (end point primario) y los parámetros hemodinámicos, la CF OMS y alivio sintomático con prolongación del intervalo de tiempo hasta el deterioro clínico (endpoints secundarios)135. El síncope se presenta en el 4% de los pacientes del grupo placebo versus el 1% de los tratados con riociguat. Está contraindicada su combinación con IPDE-5 por mayor efecto hipotensor.
3.4.5 Análogos de las prostaciclinas (prostanoides) y agonistas de los receptores de prostaciclinas
La prostaciclina es un potente vasodilatador e inhibidor de la agregación plaquetaria, tiene, además, propiedades citoprotectoras y antiproliferativas. En pacientes con HAP se ha descripto disminución de la prostaciclina sintetasa en la vasculatura pulmonar.
Beraprost. Es un prostanoide de administración oral. Su uso se ha asociado con mejoría de la capacidad de ejercicio sostenida por hasta seis meses136. Los efectos adversos más frecuentemente comunicados han sido cefalea, dolor mandibular y diarrea. Este medicamento no está aprobado ni disponible en Europa, tampoco en Argentina.
Epoprostenol. Es una solución inestable que requiere infusión endovenosa continua por bomba a través del implante de un catéter permanente. Es la única droga que demostró disminuir la mortalidad en HAPI. Se recomienda una dosis inicial de 2-4 ng/kg/min con titulación en función de la tolerancia; la dosis óptima oscila entre 20 y 40 ng/kg/min10. La suspensión brusca se asocia con riesgo de efecto rebote de HP con agravamiento sintomático y hasta muerte. En pacientes con HAPI CF OMS III-IV y HAP-ES, se observó mejoría de la capacidad de ejercicio y los parámetros hemodinámicos y alivio sintomático; además de disminucion del riesgo de mortalidad de aproximadamente el 70%137. El efecto beneficioso a largo plazo se ha evidenciado en HAPI y HAP asociada a otras patologías. Las complicaciones del tratamiento en su mayoría se relacionan con la función de la bomba de infusión, la obstrucción del catetér y la infección.
Iloprost. Iloprost es una solución químicamente estable de uso intravenoso o inhalatorio. En pacientes con HAP y HPTEC la inhalación de 6 a 9 puffs de 2.5 a 5 μg (promedio 30 μg/día) se asoció con mejoría de la capacidad de ejercicio, reduccion de la RVP y alivio sintomático138. El uso intravenoso ha mostrado resultados comparables a los de epoprostenol.
Treprostinil. Es una solución químicamente estable. Se administra por vía subcutánea mediante una bomba de microinfusión continua con una dosis inicial de 1-2 ng/kg/min oscilando el rango óptimo entre 20 y 80 ng/kg/min. El tratamiento con treprostinil se asocia a mejoría de la capacidad de ejercicio y los parámetros hemodinámicos, y alivio sintomático139, particularmente en individuos con mayor compromiso inicial y que toleran las dosis más altas (>13.8 ng/kg/min). El efecto adverso más frecuente fue dolor en sitio de la infusión con discontinuación en el 8% de los casos. También se ha aprobado el treprostinil por vía inhalatoria140, vía endovenosa141 y vía oral142.
Selexipag. Es un agonista selectivo del receptor IP de la prostaciclina de administración oral. Un ensayo clínico fase III en más de 1000 pacientes con HAP, en monoterapia o combinación con ARE o IPDE-5 reflejó una disminución del criterio de valoración compuesto (deterioro de HAP con internación, trasplante, septostomía atrial, prostanoides intravenosos u oxigenoterapia y mortalidad) del 39%143. Este medicamento, por via oral, no está aún aprobado en Argentina.
3.4.6 Monoterapia
No hay ensayos clínicos comparativos droga a droga o “head to head” ni otras evidencias que postulen a un fármaco como de primera elección. Prescribir uno u otro fármaco depende de la aprobación en el país, vía de administración, perfil de seguridad y tolerancia, interacción con otros fármacos, comorbilidades, experiencia del grupo tratante, costos de financiamiento y consenso con el paciente (Tabla 30).
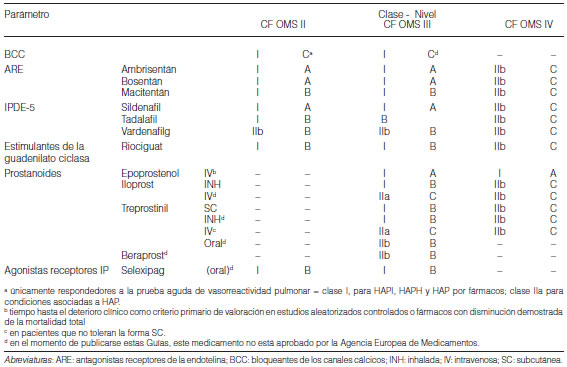
3.4.7 Terapia combinada
La terapia combinada, con dos o más fármacos, ha mostrado beneficios en el tratamiento de la HAP dado que pueden simultáneamente actuar sobre la vía de la prostaciclina, la endotelina y el ON. Un metanálisis de seis ensayos clínicos ha concluido que la terapia combinada disminuye el riesgo de deterioro clínico, mejora significativamente los resultados de la PM6M (+ 22 metros) y disminuye la PAP y la RVP, con un buen perfil de seguridad y tolerancia144. Un ensayo clínico reciente145 con terapia combinada inicial, mostró una disminucion significativa de la RVP en los tratados con la combinación epoprostenol/bosentán versus epoprostenol monoterapia, aunque este beneficio no se reflejó en la tasa de supervivencia o supervivencia libre de trasplantes. La ventaja de este abordaje es que aún los pacientes estables o aquellos con una leve mejoría pueden seguir beneficiándose del agregado de otro fármaco. El tratamiento se considera adecuado únicamente si se alcanzan dichos objetivos10.
Recientemente, un estudio multicéntrico, multinacional146 en pacientes incidentales y vírgenes de todo tipo de tratamiento farmacológico previo, comparó taladafil monoterapia versus ambrisentán monoterapia versus una combinación de taladafil/ambrisentán como estrategia de inicio, en pacientes con HAP CF OMS II-III. Se observó una disminucion del 50% de los eventos (muerte, internación, progresión, condición clínica insatisfactoria) en los tratados con la combinación versus la monoterapia cualquiera fuera ella. También se beneficiaron la capacidad de ejercicio, tasa de respuesta clínica satisfactoria y Niveles de NT-proBNP. Si bien se puede prescribir desde el inicio del tratamiento, la terapia combinada se ha implementado hasta el momento en forma secuencial, basado en el conocido esquema terapéutico guiado por metas. Las recomendaciones de las Guías ESC/ERS 2015 respecto de la terapia combinada, inicial o secuencial, se pueden apreciar en las Tablas 31 y 3210.

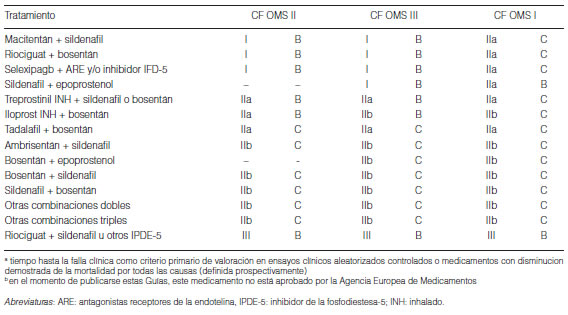
3.5 Abordaje terapéutico de las complicaciones
3.5.1 Arritmias
En pacientes con HAP la arritmia auricular (fibrilación y aleteo) sintomática es frecuente y de pobre pronóstico, ambas asocian deterioro clínico y signos de insuficiencia del VD. La restitución de un ritmo normal, más probable en el aleteo auricular, se asocia con una buena supervivencia a largo plazo; en tanto la persistencia de la fibrilación implica un riesgo de mortalidad en dos años cercano al 80%. Para mantener un ritmo sinusal estable se puede indicar amiodarona. Las arritmias supraventriculares crónicas requieren anticoagulación crónica.
3.5.2 Hemoptisis
La hemoptisis, más frecuente en pacientes con HAPH, HAP asociada a cardiopatia congénita y HPTEC, tiene una prevalencia del 1 a 6%. Puede ser tan grave que ponga en riesgo la vida y en estos casos se puede considerar la embolización de la arteria bronquial. La hemoptisis es una contraindicación para el uso de anticoagulantes.
3.5.3 Complicaciones mecánicas
La dilatación de la AP puede asociar aneurismas con ruptura y disección, y compresión de estructuras intratorácicas como el tronco de la arteria coronaria izquierda, venas pulmonares, bronquios, nervio recurrente. Clínicamente el cuadro puede manifestarse por disnea, dolor precordial, edema pulmonar y hasta muerte súbita. La angiotomografía multicorte y la TACAR es el método más confiable para detectar estas complicaciones, aunque en algunos casos, se requiere de la cinecoronariografia cuando se sospeche compresión del tronco de la coronaria izquierda. Las opciones terapéuticas van desde la colocación de un stent con acceso percutáneo al trasplante en pacientes crónicos estables, pero no hay una conducta terapéutica definida10.
3.6 Manejo de la insuficiencia ventricular derecha avanzada
3.6.1 Internación en Unidad de Terapia Intensiva
La insuficiencia del VD es una de las circunstancias en las que un paciente con HP requiere internación en la unidad de terapia intensiva (UTI) (I-C)10. Básicamente, el manejo consiste en tratar la causa subyacente (comorbilidades, arritmias, etc.), optimizar el balance de fluidos, disminuir la poscarga del VD, y mantener la presión sanguínea. El uso de inotrópicos para mejorar el gasto cardíaco está recomendado en pacientes hipotensos CF OMS III-IV (I-C)10. Un dato fundamental es que la intubación debe evitarse ya que puede precipitar un colapso hemodinámico10. Ensayos clínicos sugieren que el uso de dispositivos de soporte vital extracopóreo, como la membrana de oxigenación extracorpórea (ECMO, extracorporeal membrane oxygenation) veno-venoso o veno-arterial, pueden ser una opción para pacientes en estadios muy avanzados de insuficiencia del VD que están en lista de espera de trasplante, como puente hasta su realización147. Preferentemente se recomienda el ingreso a una UTI en un centro de referencia, teniendo en cuenta que se debe privilegiar el criterio clínico del medico tratante (Tabla 33)10.

3.6.2 Septostomía auricular con balón
Esta técnica crea un cortocircuito derecha/izquierda que contribuye a disminuir la presión de la AD, descomprimir el VD y aumentar la precarga izquierda y el VM, mejorando el transporte tisular de O2, independientemente de la desaturación arterial procada por el procedimiento. Además disminuye la actividad simpática, observándose mejoría del IC y la PM6M, y disminución de la presión arterial diastólica. La intervención se realiza a nivel del foramen oval mediante un catéter con balón el cual se dilata gradualmente. Esto produce una mejoría similar en la hemodinamia y los síntomas10, 125. La mortalidad de la septostomía auricular en manos de expertos es de 7,1% y 14,8% a las 24 horas y 30 días148. Este procedimiento es considerado como un paliativo y debe realizarse en centros con amplia experiencia5, 10, 125. Se puede considerar en pacientes CF OMS III-IV cuando ha fracasado la terapia médica máxima (IIb-C), aunque no se incluye en el algoritmo de tratamiento de las Guías ESC/ERS 2015 (Tablas 34 y 35)10. Recientemente un estudio retrospectivo del uso de la septostomía auricular asociada al uso de fármacos específicos mostró una mortalidad del 2%149.


3.7 Trasplante pulmonar y cardiopulmonar
En pacientes que persisten en CF III –IV con mala respuesta al tratamiento farmacológico máximo, el trasplante pulmonar bilateral o el cardiopulmonar es una opción razonable (I-C)10, 125. Según el registro de la International Society for Heart y el Lung Transplantation en la actualidad predomina la tendencia al trasplante bipulmonar150. Esta misma sociedad ha publicado en enero de 2015 un documento de consenso generado por un comité de expertos para la selección de candidatos para trasplante pulmonar151 donde se establecen criterios clínicos para la derivación y para inclusión en lista de espera para trasplante de pacientes con enfermedad vascular pulmonar. El pronóstico de las distintas etiologías de la HAP contribuye a la decisión terapéutica. El pronóstico de la HAP-ETC es peor que el de la HAPI, en tanto la mejor supervivencia es la de los pacientes con HAP asociada a cardiopatías congénitas. En el síndrome de Eisenmenger es más difícil establecer el momento para ingreso en lista de espera, porque la supervivencia es mayor librada a la evolución natural de la enfermedad5.
En cualquier caso, los pacientes deben ser derivados tempranamente antes de la aparición de insuficiencia renal, cardíaca o hepática, porque estas entidades aumentan la mortalidad en lista de espera. Datos recientes muestran una tasa de supervivencia de 52 a 75% a los 5 años y de 45 a 66% a los 10 años152-154. En la EPVO y la HCP la terapia farmacológica es inefectiva y los pacientes deben ser derivados para trasplante en el momento del diagnóstico10. En las Tablas 36 y 37 se detallan las Disposiciones 2010 del INCUCAI relacionadas con las distintas situaciones clínicas para trasplante pulmonar (uni o bipulmonar) y cardiopulmonar y, los criterios de asignación de órganos intratorácicos para órganos provenientes de donantes cadavéricos155. En la Tabla 38 se describen los criterios de abordaje terapéutico de los pacientes candidatos a trasplante con insuficiencia del VD.



3.8 Algoritmo terapéutico
Más allá de la clase de recomendación y nivel de evidencia, la realidad indica que el abordaje terapéutico de la HAP depende de las regulaciones y disponibilidad local de medicamentos. El algoritmo que proponen las Guías ESC/ERS 2015 aplica únicamente para la HAP Grupo 1, con la particularidad de haber sido evaluado principalmente en individuos con HAPI, HAPH, HAT-ETC, HAPD y HAP con cardiopatía congénita (corregida o no quirúrgicamente). Están fuera del alcance de este algoritmo los Grupos 2 y 3 de HP10 (Figura 2).
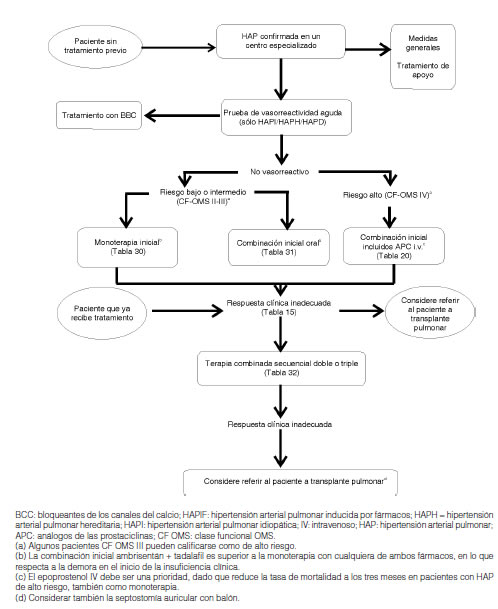
Consideraciones
– Si la PVRPA es positiva iniciar tratamiento con altas dosis de BCC, se debe reevaluar la respuesta a los 3-4 meses.
– Si la PVRPA es negativa, con criterios de riesgo bajo o intermedio se recomienda iniciar tratamiento con monoterapia o terapia combinada.
– No es posible recomendar una monoterapia de primera línea. Los criterios de selección del fármaco son los que se describen más arriba (Tabla 30).
– En cuanto a la terapia combinada: hay evidencia documentada de la superioridad de ambrisentán/ taladafil versus monoterapia con ambrisentán o taladafil.
– Si la PVRPA es negativa o el paciente es vírgen de tratamiento, con criterios de alto riesgo, la terapia combinada inicial debe incluir análogos de la prostaciclina (epoprostenol EV). Pueden utilizarse combinaciones alternativas.
– Si la respuesta es inadecuada al tratamiento inicial (monoterapia o combinación) puede considerarse terapia secuencial. Recordar la contraindicación de asociar riociguat con IPDE-5.
– Ante una respuesta inadecuada al tratamiento combinado máximo: se recomienda derivar a un centro especializado en trasplantes.
– En caso de haber un rápido deterioro: la septostomía auricular con balón puede ser un paliativo hasta el trasplante, si no existe contraindicación.
3.9 Ética y cuidados al final de la vida
La evolución clínica de la HP se caracteriza por un deterioro progresivo con episodios de descompensación intercurrentes. La muerte puede acontecer súbitamente o como consecuencia de la insuficiencia cardiaca derecha progresiva. Esto implica, para el equipo tratante, la responsabilidad de discutir, con honestidad y realismo, las opciones que el paciente y su familia prefieren para el fin de la vida. Se recomienda crear una situación, preferentemente al inicio del diagnóstico, para trasmitir la verdad sobre la enfermedad y su pronóstico, con el bjetivo de permitir al paciente tomar decisiones esenciales sobre su propia vida como la aceptación o rechazo de medidas terapéuticas heroicas (ventilación asistida, resucitación cardiopulmonar) ante situaciones de extrema gravedad10. Es función del médico no faltar a la verdad y, al mismo tiempo, lograr un apoyo firme en el difícil proceso de toma de decisiones. Un equipo multidisciplinario con experiencia en cuidados paliativos facilitará este proceso, pero el médico tratante tiene la responsabilidad indelegable de discutir estas decisiones con el paciente y su familia5, 156, 157.
4. Hipertensión arterial pulmonar específica
4.1 HAP en pediatría
4.1.1 Definición
La HP se define en los niños por la presencia de valores de PAPm mayores o iguales a 25 mmHg medida a partir de los tres meses de edad, en un niño nacido a término, en presencia de una distribución regular del flujo sanguíneo en todos los segmentos pulmonares158. Esta definición no distingue entre pacientes con o sin enfermedad vascular pulmonar hipertensiva. En los niños los tipos más frecuentes de HP son la HAPI y las HP asociada a cardiopatías congénitas (HP-CC).
4.1.2 Clasificación
La clasificación de Niza hace foco en varios aspectos, se añadieron algunos elementos específicos relacionados con la HP pediátrica con el fin de tener una clasificación exhaustiva común para adultos y niños8,10. Dada su particular anatomía y fisiología, la HP persistente del recién nacido (persistencia del patrón fetal) fue clasificada como una subcategoría del Grupo 1, remarcando los aspectos específicos: inicio inmediatamente después del nacimiento, evolución aguda, y las estrategias terapéuticas158. Asimismo, se menciona la incorporación de las lesiones obstructivas, congénitas o adquiridas, de entrada y salida del corazón izquierdo y las cardiomiopatías congénitas en el Grupo 2; y de las enfermedades del desarrollo pulmonar en el Grupo 3. También se ha incluido en el Grupo 5 la categoría de HP segmentaria, y se ha modificado la clasificación respecto de la HP-CC (Tabla 39)8, 10, 158. Asimismo, se reconocieron lesiones en las cuales el compromiso de la vasculatura pulmonar es posible aunque sin superar los 25 mmHg de PAPm es decir que no cumplen con los criterios específicos de HP por lo cual no se los ha incluido en esta clasificación. Esto incluye pacientes con fisiología de ventrículo único quienes deben recibir una corrección oximétrica (Glenn bidireccional ó bypass de VD “Técnica de Fontan–Kreutzer”); en esta cirugía con flujo no pulsátil a las arterias pulmonares la PAP no puede superar los 25 mmHg; sin embargo, la existencia de un grado significativo de enfermedad vascular pulmonar puede producir pobres resultados158.

4.1.3 Cardiopatías congénitas
Los defectos cardíacos congénitos ocurren en el 10‰ de los recién nacidos vivos; de éstos, entre el 50-60%, son casos de cortocircuitos sistémico-pulmonares, y aproximadamente un 5-10% de estos pacientes, presentarán HAP en la edad adulta5, 159. La probabilidad de desarrollar enfermedad depende del tamaño y localización del defecto. Se produce en el 10% en la comunicación interauricular, en el 3% en la comunicación interventricular mediana (<1 cm de diámetro) y en el 50% en la comunicación interventricular grande (>1 cm de diámetro), en el 100% con tronco arterioso y en el 50% en el ductus arterioso permeable5, 159.
La Clasificación de Niza también se ha modificado con respecto a la HP asociada con CC158.
– Tipo 1 Síndrome de Eisenmenger: es la forma más avanzada de HAP-CC, en el que la persistente exposición del lecho vascular a un incremento del flujo sanguíneo conduce a un aumento en la presión pulmonar produciendo una arteriopatía obstructiva pulmonar. Las resistencias pulmonares se elevan a nivel sistémico o suprasistémico y se invierte el cortocircuito de derecha a izquierda generando desaturación sistémica (cianosis central). Incluye los grandes defectos intra y extracardíacos que se inician como un cortocircuito desde la circulación sistémica a la pulmonar y evolucionan al aumento importante de la RVP y cianosis por cortocircuito reverso o bidireccional, asociándose usualmente a eritrosis secundaria y compromiso multiorgánico5, 158.
– Tipo 2 Cortocircuito izquierda-derecha: corregible o no corregible. Incluye defectos moderados a graves, aún prevalece un cortocircuito entre la circulación sistémica y la pulmonar; la cianosis no es una característica. Incluye pacientes con CC y significativa enfermedad vascular hipertensiva pulmonar con normal saturometría en reposo. Los cortocircuitos pueden ser corregibles o no, pero se caracterizan por aumento leve a moderado de la RVP
– Tipo 3 HAP coincidente con pequeños defectos: se caracteriza por un marcado aumento de la RVP en presencia de defectos cardíacos pequeños que no justifican per se el aumento de la RVP. Se contraindica el cierre de estos defectos. El curso clínico y la evolución son similares a la HAPI.
– Tipo 4 HAP post corrección quirúrgica: puede ser una enfermedad persistente después de reparar una cardiopatía congénita o desarrollarse meses o años más tarde, aún en ausencia de lesiones hemodinámicas significativas. El fenotipo clínico suele ser agresivo. Incluye pacientes con algún tipo de CC corregida quienes desarrollaron enfermedad vascular pulmonar hipertensiva158.
4.1.4 Diagnóstico
Un enfoque diagnóstico metódico y exhaustivo es importante porque son muchas las enfermedades asociadas con HP.
PVRPA: la respuesta a esta prueba es fundamental para determinar inicialmente la atención del niño (Tabla 40). No hay droga estándar para el PVRPA en pediatría; pero el ON inhalado (en dosis de 20 0 80 partes por millón) es el más frecuentemente utilizado y se aconseja para éste propósito. En cuanto a los parámetros para medir resultados los más utilizados son los criterios de Barst modificados (Tabla 41)160, aunque en la evaluación de la corrección de una CC, no hay establecido un protocolo de PVRPA que proporcione criterios de respuesta o de seguimiento a largo plazo.


En la Figura 3 se refleja el algoritmo relativo al procedimiento diagnóstico modificado propuesto en Niza158.
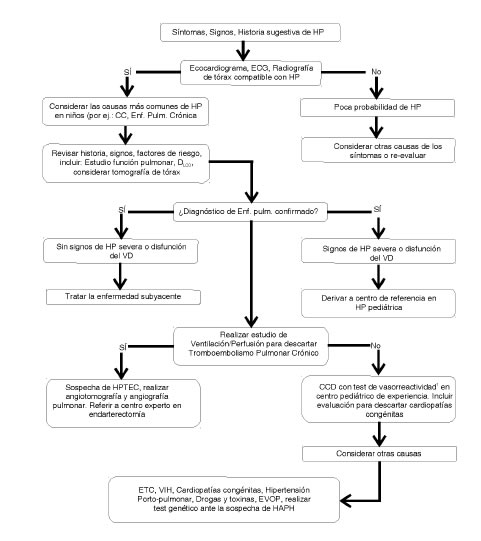
4.1.5 Seguimiento - Parámetros para el control del tratamiento
Los parámetros que miden cómo se siente, qué actividad realiza y cuál es la supervivencia de un paciente incluyen la CF y la prueba de esfuerzo. Si bien la CF no ha sido diseñada específicamente para los niños, ha mostrado correlación con la prueba de marcha y parámetros hemodinámicos161. Aún no ha sido validado, pero un diseño específico de CF para niños ha sido propuesto162. Los resultados de diversas evaluaciones permiten determinar el riesgo de mortalidad de los pacientes, esencial para orientar el manejo terapéutico: CF-OMS III/IV y niveles aumentados de BNP, indicios de insuficiencia VD y progresión de los síntomas son indicadores de alto riesgo de mortalidad. También se asocian con alto riesgo el retraso del crecimiento, presión auricular derecha > 10 mmHg e índice de RVP > 20 UW/m2 (Tabla 42)10, 158. En estudios de seguimiento, la distancia recorrida en la PM6M no fue un predictor de supervivencia10, 163. En el CCD durante el seguimiento la constancia de una PVRPA positiva se asocia con una mayor supervivencia. Se recomienda repetir este estudio cuando el paciente presente deterioro clínico; para evaluar el efecto del tratamiento; para la detección temprana del deterioro de la enfermedad; para valorar la indicación de trasplante; para la predicción de pronóstico160. Una prueba no invasiva también utilizada para el seguimiento es el ecocardiograma (índice de excentricidad, excursión del plano del anillo tricuspídeo, derrame pericárdico, Doppler tisular).
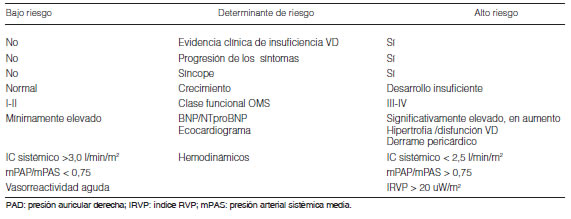
4.1.6 Tratamiento
El objetivo del tratamiento es mejorar la supervivencia y lograr que el niño realice una actividad acorde a su edad, sin necesidad de autolimitarse. La terapia con oxígeno, digoxina, diuréticos ó anticoagulantes deben ser evaluadas en cada paciente.
4.1.6.1 Bloqueantes cálcicos
En los pacientes mayores de un año de edad con PVRPA positiva el tratamiento puede iniciarse con BCC (nifedipina - amlodipina - diltiazem)164; si el niño mejora y sostiene la respuesta se continúa con esta indicación, pero ante signos ó síntomas de deterioro o PVRPA negativa se debe replanificar el tratamiento.
4.1.6.2 Monoterapia
En los niños con bajo riesgo y PVRPA negativa el tratamiento recomendado es monoterapia por vía oral con ARE (bosentán, ambrisentán) o IPDE-5 (sildenafil, taladafil). Ensayos clínicos con bosentán han mostrado supervivencia del 80 a 90% al año, existiendo en Europa una formulación pediátrica10. Los análisis realizados por la FDA (Food and Drug Administration) y por la EMA (European Medicines Agency) de los resultados de los ensayos clínicos STARTS-1 y STARTS-2 (Sildenafil in Treatment-Naive Children) en pacientes de 1 a 17 años, con HAP) resultaron en recomendaciones dispares. Fue aprobado para su uso por la EMA en 2011 siendo las dosis máximas 10 mg/dosis en niños con un peso entre 8 y 20 kg; 20 mg/ dosis en aquellos con más 20 kg o >1 mg/kg/dosis en bebes y niños pequeños10. En tanto en 2012 la FDA emitió un alerta sobre el uso crónico de la droga en altas dosis, que posteriormente rectificó admitiendo su utilidad pero no recomendándolo. En cuanto al epoprostenol, las dosis requieren adaptación individual10.
4.1.6.3 Terapia combinada
Los pacientes que deterioran su condición clínica con la monoterapia pueden beneficiarse de una terapia combinada. Si permanece en la categoría de bajo riesgo puede adicionarse una prostaciclina inhalada. En niños en la categoría de alto riesgo debe plantearse una prostaciclina subcutánea ó endovenosa.
4.1.6.4 Cirugía de descompresión del VD
La septostomía auricular es un procedimiento de alto riesgo en estadios avanzados con alta presión en la AD y bajo VM. Ofrece una opción en pacientes con rápido empeoramiento a pesar del tratamiento farmacológico adecuado y en lista de espera para trasplante. En pacientes con PAPm suprasistémica ha sido propuesta o la creación quirúrgica ó endovascular de un cortocircuito entre la aorta descendente y la rama pulmonar izquierda (Potts) En pacientes con ductus arteriosus permeable se puede considerar la colocación de un stent (endoprótesis) ductal10, 158.
4.1.6.5 Trasplante pulmonar
Un punto muy importante en el manejo a largo plazo de los pacientes con HP es la evaluación periódica de la respuesta a las terapias específicas y la progresión de la enfermedad. En aquellos con deterioro rápido se debe considerar la posibilidad de un trasplante bipulmonar.
4.1.7 Consideraciones finales
El pronóstico, la calidad de vida y la sobrevida en los niños con HP han mejorado en la última década gracias a los nuevos agentes terapéuticos y estrategias de tratamiento agresivo. Una etiología compleja y la falta de datos en este grupo etario hace difícil la selección de terapias apropiadas las cuales se basan, fundamentalmente, aún en los resultados de ensayos clínicos en adultos y en la opinión de expertos (Figura 4)158. Finalmente, en la tabla se resumen las recomendaciones de las Guías 2015 de las sociedades ESC/ERS relativas al manejo de la HAP en pediatría (Tabla 43)10.
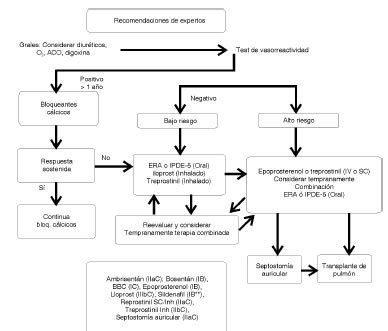

4.2 HAP asociada a enfermedad del tejido conectivo
Luego de la HAPI, la HAP asociada a ETC (HAP-ETC) es la más frecuente en los registros, siendo la ES limitada la más preponderante con una prevalencia de 7-12%5, 165. Le siguen en frecuencia la forma difusa de la ES y la EMTC, aunque la HAP puede presentarse en cualquier otra ETC, como LES, miopatías inflamatorias, AR, síndrome de Sjögren, síndrome antisintetasa, etc. La HAP puede ocurrir como resultado de una enfermedad vascular aislada con compromiso de las arteriolas precapilares y vénulas poscapilares, o asociada a una enfermedad pulmonar intersticial. También estos pacientes pueden desarrollar HP del Grupo 2 (enfermedad cardiaca izquierda). Es esencial distinguirlas porque el tratamiento y pronóstico son diferentes. Los pacientes con HAP-ETC tienen menor supervivencia que aquellos con HAPI5, 10, 166, 167. La TACAR descarta enfermedades del intersticio pulmonar y EPVO. La disminución aislada de la DLCO en ausencia de compromiso intersticial en la TC o con volúmenes pulmonares normales implica la necesidad de descartar HAP2, 3, 5. El perfil inmunológico contribuye a la identificación del tipo de ETC (Tabla 44).

4.2.1 Consideraciones diagnósticas
– En pacientes con ETC comprendidas en el espectro de la ES sin signos ni síntomas cardiopulmonares, es conveniente un examen anual para detectar HAP(9) Se recomienda un ecocardiograma en reposo, DLCO y biomarcadores (I-C)10.
– En caso de sospecha de HAP-ETC (PAPS > 40 mmHg y/o la velocidad de reflujo tricuspídeo ≥ 2,8 m/s), se sugiere realizar un CCD para confirmar HAP (I-C)5, 10.
– En pacientes con sospecha de HAP-ETC está indicado el CCD para confirmar el diagnóstico y evaluar la severidad de la enfermedad9 (I-C)10.
– Aquellos con PAPm entre 21-24 mmHg se encuentran en alto riesgo de desarrollar HP en el término de tres años y se recomienda monitoreo periódico168.
4.2.2 Consideraciones terapéuticas
– El tratamiento de la HAP-ETC es idéntico al de la HAPI, con buena respuesta, en general, a las terapias especificas (I-C)10.
– Considerar anticoagulación oral individualizada en presencia de trombofilia (IIb-C)10.
– En pacientes con ES la prescripción del tratamiento debe tener presente la microangiopatía asociada, siendo relevante la investigación de telangiectasias intestinales mediante la realización de endoscopía, generalmente se trata de lesiones limitadas que se manifiestan clínicamente por hemorragia digestiva baja169.
– La respuesta a la PRVPA suele ser negativa y, cuando el paciente es respondedor, la eficacia de los BCC se extingue a lo largo del tiempo170. En aquellos casos con evidencia de inflamación sistémica (LES, EMTC, etc.) el uso de corticoides e inmunosupresores (ciclofosfamida) puede ser efectivo; el trabajo de Sánchez y cols (Chest 2006) revisado por la Joint Task Force establece que sólo el 50% de 23 pacientes respondió a terapia inmunosupresora más corticoides. En casos graves, se recomienda la terapia debe ser específica para HAP incluyendo prostaciclinas sistémicas10.
4.3 HAP asociada a infección por el virus de la inmunodeficiencia humana
La prevalencia de HAP en pacientes con VIH se ha mantenido estable respecto de la era preHAART (High Active Antiretroviral Therapy) (0.1-0.5%), con una incidencia estimada de 0.1% por año32. Se encontró una mayor frecuencia en el sexo masculino y usuarios de drogas endovenosas. La patogénesis podría estar relacionada con un efecto inflamatorio, aunque puede haber otros factores de riesgo como enfermedad hepática (hepatitis crónica B o C), exposición a drogas o embolismo pulmonar en los usuarios de drogas endovenosas5. A partir del advenimiento de la HAART y las drogas especificas para la HAP el pronóstico ha mejorado notoriamente; la tasa anual de mortalidad del 50% se ha transformado en una tasa de sobrevida del 70% a los 5 años8, 171, y hasta se ha informado normalización de los parámetros hemodinámicas después de varios años de tratamiento en un 20% de los casos8, 172.
4.3.1 Consideraciones diagnóstico terapéuticas
– No se recomienda la realización sistemática de ecocardiograma en pacientes asintomáticos (III-C)10. No obstante, este examen está indicado en pacientes VIH positivos con disnea para detectar complicaciones cardiovasculares y, en ausencia de estos eventos, determinar sospecha de HAP-VIH5, 10, 32.
– El tratamiento es el mismo que el de la HAPI más la HAART, considerando las comorbilidades e interacciones con el tratamiento antirretroviral (IIa-C)5, 10, 173.
– La anticoagulación asocia riesgo de sangrado e interacciones con la HAART; no se recomienda como tratamiento de rutina, dada la ausencia de datos respecto del cociente eficacia/riesgo (III-C)5,10.
– Estos pacientes tienen una respuesta negativa en la PVRPA; por lo tanto no se recomienda realizarla ni prescribir BCC.
– La condición VIH (+) es un criterio de contraindicación relativa para trasplante ya que se lo está implementando en algunos centros.
4.4 HAP asociada con hipertensión portal (hipertensión portopulmonar)
Actualmente, se estima que entre el 1% y 5% de los pacientes con hipertensión portal (HPo) desarrollan HAP configurando la condición clínica conocida como hipertensión portopulmonar (HPoP)166. Su definición es hemodinámica con los criterios clínico o hemodinámico de HAP asociada a la clínica o hemodinamia de presencia de hipertensión portal. En el Registro Francés, esto representó, aproximadamente, el 10% de todas las formas de HAP12. Las hipótesis acerca de la patogénesis involucra a sustancias tóxicas provenientes del tracto gastrointestinal que provocan daño endotelial en el lecho vascular pulmonar y riesgo genético2, 3, 10. El riesgo de desarrollar HPoP es independiente de la gravedad del compromiso hepático y de la severidad de la HPo. En contraste, la gravedad de la cirrosis y la insuficiencia cardiaca determinan el pronóstico a largo plazo174.
4.4.1 Consideraciones diagnósticas
En general, los signos y síntomas son similares a los de HAPI, con el agregado, en algunos pacientes, de fenómenos asociados a la hipertensión portal (circulación colateral, ascitis, etc.)175. Un dato importante es que se puede encontrar HP en presencia de cirrosis sin que esto constituya una HPoP dado que la HP puede estar asociada a sobrecarga de volumen, aumento de la presión de fin de diástole del VI o a el elevado VM que presentan las hepatopatías.
En los pacientes con hipertensión portal o enfermedad hepática y disnea de esfuerzo no atribuible a otra causa, y en todos aquellos pacientes candidatos a trasplante hepático el método de elección de cribado es el ecocardiograma Doppler para descartar la presencia de HP (I-B)10. La hipertensión portopulmonar es de mal pronóstico y condiciona la posibilidad de recibir un trasplante hepático2, 3, 5, 10, 29.
4.4.2 Consideraciones terapéuticas
– Estos pacientes deben ser derivados a centros de referencia con experiencia en el manejo de ambas condiciones (hipertensión portal y HAP) (I-C)10.
– El abordaje terapéutico de la HPoP no difiere del de las otras formas de HAP aunque debe tenerse en cuenta la gravedad del compromiso de la función hepática (I-C)2, 3, 10.
– La anticoagulación está limitada por los trastornos espontáneos de la coagulación y no está recomendada (Nivel de recomendación III, clase C)2, 3, 10.
– La presencia de hipertensión portopulmonar determina un marcado aumento de la mortalidad para los pacientes candidatos a trasplante hepático; hasta el 100% en pacientes con PAPm ≥ 50 mmHg y 50% en aquellos con PAPm entre 35 and 50 mmHg y RVP ≥ 250 dyn.s.cm-5. Se puede considerar el trasplante en pacientes seleccionados con buena respuesta al tratamiento de la HAP que presenten una PAPm < 35 mmHg (IIb-C)10. En cambio, está contraindicado en individuos con HAP grave no controlada (III-C)10. Como último recurso y en enfermos altamente seleccionados, existe la posibilidad de realizar la combinación de trasplante hepático y trasplante pulmonar (o cardiopulmonar), con tasas de sobrevida del 49% a cinco años en centros especializados2, 3, 10.
– También se recomienda evitar el uso de betabloqueantes10, 176. Algunas series mostraron la eficacia de IPDE-5, AREs y análogos de la prostaciclina y riociguat, con mejoría de la hemodinamia y los síntomas10, 177. El uso de bosentán requiere cuidadosa evaluación del grado de disfunción hepática, debido a su potencial toxicidad175.
4.5 HAP asociada con enfermedad cardiaca congénita en el adulto
La HAP es frecuente en los pacientes adultos con enfermedad cardíaca congénita y se incluye en el Grupo 1 de la HP10. El Registro Francés demostró que las cardiopatías congénitas representaron el 11.3% de todas las formas clínicas de HAP12; el registro CONCOR (Holanda) reveló que el 4.2% de 5970 pacientes adultos con cardiopatías congénitas tenían HAP37. La población que conforma este grupo es heterogénea. El aumento de la RVP se explica por la exposición de los vasos pulmonares a un aumento del flujo sanguíneo como consecuencia de los cortocircuitos sistémico-pulmonares. El diagnóstico requiere la búsqueda minuciosa de defectos tales como el ductus arterioso persistente, defectos del septum venoso atrial o retorno venoso anómalo, los cuales suelen pasar desapercibidos llevando al diagnóstico erróneo de HAPI.
4.5.1 Consideraciones pronósticas
Respecto de la evolución se aclara que aquellos en quienes el cortocircuito no revierte cursan con un grado leve a moderado de cianosis y eritrosis. La supervivencia depende, fundamentalmente, de la conservación de la función del VD; y está más comprometida en aquellos con reparación del defecto o la coexistencia de varios pequeños defectos en comparación con el Síndrome de Eisenmenger y los individuos con cortocircuito sistémico-pulmonar; también estos pacientes pueden desarrollar HP del Grupo 2 (enfermedad cardiaca izquierda), o del Grupo 3 (enfermedad pulmonar).
4.5.2 Abordaje Terapéutico
4.5.2.1 Manejo quirúrgico
La cirugía es la primera opción en pacientes con cortocircuito sistémico-pulmonar (Tabla 45).

4.5.2.2 Abordaje farmacológico
El tratamiento medicamentoso de la HAP asociada a enfermedad cardiaca congénita (Tabla 46) debe incluir, además, educación del paciente, modificaciones conductuales y advertencias sobre riesgos médicos. Diversas circunstancias pueden precipitar el deterioro clínico: cirugías con anestesia general, deshidratación, infección pulmonar, altitud, actividad física extenuante. Se desaconseja el embarazo ya que implica un gran riesgo maternofetal.

4.6 HAP asociada con esquistosomiasis
En las nuevas guías ESC/ERS 2015, si bien se incluye la esquistosomiasis en la clasificación del la HAP (Grupo 1), únicamente se advierte de la necesidad de investigar su presencia en las regiones donde las enfermedad es endémica10. El impacto epidemiológico de la HAP asociada con esquistosomiasis queda reflejada en un estudio brasilero del cual surge que el 19.7% de 178 nuevos casos de HAP se asociaban a esquistosomiasis22.
4.7 Enfermedad pulmonar veno oclusiva y hemangiomatosis capilar pulmonar - Grupo 1’
La EVOP y la HCP son condiciones poco frecuentes; cuyas características patológicas, clínicas y riesgo de edema pulmonar inducido por los fármacos específicos para tratar la HAP se superponen. Esta particularidad sugiere que la HCP podría ser secundaria a un fenómeno angioproliferativo causado por la obstrucción poscapilar de la EVOP. Estas similitudes las han colocado en el mismo Grupo 1’, de la clasificación de HAP. La EVOP y la HCP pueden ser idiopáticas, asociadas a mutaciones y, también, complicar otras condiciones mórbidas. La proporción de EVOP/HCP idiopáticas oscila en el 10%; predomina en varones y su pronóstico es peor que el de la HAPI (Ver apartado 1 de la Tabla 4).
4.7.1 Consideraciones diagnósticas
– El diagnóstico de EVOP y HCP requiere la combinación de signo sintomatología clínica (disnea de ejercicio y fatiga), hallazgos en el examen físico (hipocratismo digital, crepitaciones bibasales) y estudios complementarios (hipoxemia, disminucion DLCO)5, 96, 178.
– También son relevantes la broncoscopía y el estudio del fluido del lavado broncoalveolar (macrófagos cargados de hemosiderina). La TACAR es de elección, engrosamiento de las líneas septales subpleurales, opacidad en vidrio esmerilado centro lobulillar y linfadenopatía mediastinal se encuentran en el 100% de EVOP/HCP y se correlacionan con el riesgo de edema pulmonar con el uso de vasodilatadores (I-C)10.
– Los casos familiares de EVOP/HCP se presentan, por lo general, en hermanos jóvenes con padres sanos lo que sugiere transmisión recesiva. Se recomienda confirmar el diagnóstico investigando la presencia de mutaciones bialélicas del gen EIF2AK4. (I-B)10.
– Con las conductas anteriores se evita la biopsia quirúrgica (estándar de oro) cuyo riesgo operatorio y posoperatorio es muy elevado y por ello no recomendable5, 10, 96, 179, 180.
– Se recomienda derivar a los potenciales candidatos a trasplante a un centro de referencia para ser evaluados, tan pronto como se realiza el diagnóstico (I-C)10.
– Pruebas hemodinámica. La PAPE es normal dado que las alteraciones se presentan en pequeñas vénulas y capilares y no afectan a las grandes venas pulmonares. La realización de la PVRPA conlleva riesgo de edema pulmonar10.
4.7.2 Consideraciones terapéuticas
– Estos pacientes deben ser tratados en centros de referencia dado que están expuestos a un alto riesgo de edema pulmonar luego del inicio de la terapia específica para HAP (IIa-C)10.
– No hay comunicaciones de ensayos clínicos con evidencias concluyentes que avalen el uso de ARE, prostanoides o IPDE-5.
– El epoprostenol se asocia a edema pulmonar.
– El único tratamiento con posibilidades de curación es el trasplante pulmonar.
5. Hipertensión pulmonar por enfermedad cardíaca izquierda - Grupo 2
El aumento de conocimientos acerca de la HP ha cambiado el perfil de los pacientes con sospecha diagnóstica en quienes se solicita evaluación. En comparación con la HAP los pacientes con HP asociada a enfermedad cardiaca izquierda (HP-ECI), en particular aquellos con fracción de eyección conservada, suelen ser de más edad, prevalecen las mujeres, y son frecuentes los antecedentes de hipertensión arterial y el síndrome metabólico10, 91. Las cardiopatías izquierdas, la insuficiencia cardíaca con función sistólica preservada o disminuida y las valvulopatías son las causas más frecuentes de HP-ECI (Ver Grupo 2 de la Tabla 4). Existen amplias diferencias en los informes de prevalencia pero se estima que aproximadamente el 60% de los pacientes con insuficiencia sistólica grave y el 70% de pacientes con insuficiencia diastólica aislada padecen HP que deteriora su capacidad funcional y se asocia a mayor mortalidad91, 181.
5.1 Definición
Se han cuestionado, en el Consenso de Niza, varios aspectos relativos a la definición tradicional de la HP-ECI (PAPm ≥ 25 mmHg, PAPE > 15 mmHg y GC normal o disminuido) y, también, el uso del GPTP para distinguir entre HP pasiva y reactiva (< 12 mmHg y ≥ 12 mmHg, respectivamente)2, 3. Al respecto plantearon que se trata de una terminología poco satisfactoria que ha motivado el uso de la expresión “HP-ECI desproporcionada” con la cual se caracterizan pacientes con cambios significativos en la circulación pulmonar. Tras un cuidadoso análisis de los mecanismos fisiopatológicos se consideró esta situación como una variable hemodinámica caracterizada por ser un marcador de enfermedad91. La clave está en cómo describir la enfermedad pulmonar vascular o remodelamiento precapilar, y el remodelamiento poscapilar, y distinguirlas de un fenómeno inicialmente pasivo, es decir cómo mensurar
un cambio en la circulación pulmonar independiente de la PAPE y medible por CCD. En el Consenso de Niza se sugiere que el GDTP es el mejor parámetro para definir HP-ECI. El GPD en individuos normales oscila entre 1 y 3 mmHg y es mayor de 5 mmHg en pacientes con ECI (con excepción de los cortocircuitos)10, 91. Tanto el Consenso de Niza como las Guías ESC/ERS 2015 recomiendan, en coincidencia con la definición de HP, usar una combinación de GDTP y RVP para definir los distintos tipos de HP-ECI (p.ej.: aislada poscapilar, combinada poscapilar/precapilar) (Tabla 47)10, 91. El valor de corte de PAPE para HP precapilar debe continuar siendo ≤ 15 mmHg ya que es el que se ha usado en los ensayos clínicos que generan evidencias sobre la seguridad y eficacia de las terapias especificas.

5.2 Fisiopatología
En líneas generales, el aumento de las presiones pulmonares obedece al incremento de las presiones de llenado del VI, que lentamente generan cambios histopatológicos en la vasculatura pulmonar5.
La circulación pulmonar normal se caracteriza por presión y resistencias bajas y una gran distensibilidad de su lecho vascular. Estas condiciones permiten la adaptación a los cambios del flujo sanguíneo y el mantenimiento de un GTPD en valores de 5 a 7 mmHg sin una repercusión importante en las presiones pulmonares. Ante la persistencia de elevadas presiones de llenado ventricular izquierdo, se producen cambios en la vasculatura pulmonar. En un principio, son reversibles (HP reactiva) y responden al tratamiento vasodilatador, pero luego se establece una HP fija con cambios histopatológicos característicos (gran hipertrofia de la media y fibrosis excéntrica de la íntima). La activación neurohormonal propia de la insuficiencia cardíaca aumenta la producción global y pulmonar de citoquinas vasoconstrictoras y disminuye la producción de óxido nítrico vasodilatador (Figura 5)5, 91.
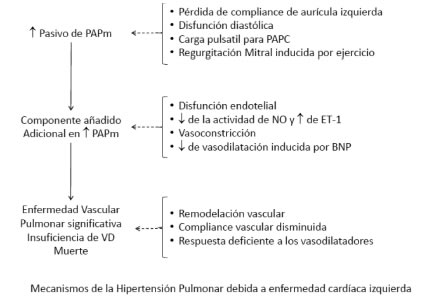
5.3 Diagnóstico
El ecocardiograma Doppler permite valorar la función sistólica y diastólica del VI y también determinar la presencia de valvulopatías. La insuficiencia cardíaca izquierda con función sistólica preservada suele ofrecer mayores dificultades diagnósticas a la hora de interpretar las imágenes ecocardiográficas5. En el CCD entre el 10% y 20% de los pacientes presentan HP combinada pre y pos capilar caracterizado por un GPDTP ≥ 7 mmHg10. Si son candidatos a trasplante cardíaco debe descartarse la HP fija. La persistencia de una RVP > 5 UW y/o GTP >15 mmHg, luego de la administración de vasodilatadores e inodilatadores, predice mayor mortalidad posoperatoria. No se ha estandarizado qué drogas deben utilizarse para esta prueba182. Las pruebas de desafío con volumen o ejercicio podrían desenmascarar una cardiopatía izquierda pero no están estandarizadas9, 10. El valor de corte de PAPE para HP precapilar debe continuar siendo ≤ 15 mmHg ya que es el que se ha usado en los ensayos clínicos que generan evidencias sobre la seguridad y eficacia de las terapias especificas. Actualmente, no hay evidencias que sustenten la realización de un CCI en la totalidad de los pacientes. Este examen debe situarse en contexto clínico y ecocardiográfico respecto de la probable existencia de enfermedad cardiaca izquierda9. En la Tabla 48 se describen los parámetros sospechosos de HP Grupo 2.
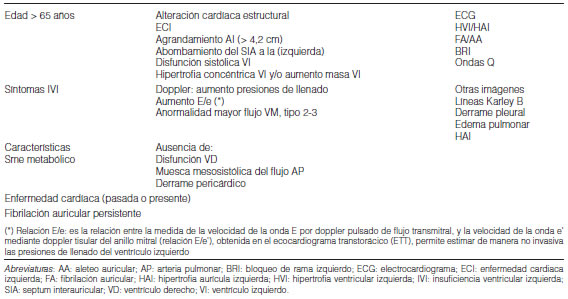
5.4 Tratamiento
Se debe mejorar la condición cardiaca subyacente antes de implementar medidas específicas para la HP5, 10, 91, 184 (Tabla 49).

La presencia de HP no contraindica ninguna droga de las actualmente recomendadas en el tratamiento de la insuficiencia cardíaca5, 182. El uso de prostanoides, IPDE-5 y AREs no está recomendado, ni existe ninguna medicación aprobada para el uso específico en HP-ECI (Tabla 50)5, 10, 91, 182, 185.

6. Hipertensión pulmonar por enfermedad pulmonar y/o hipoxia - Grupo 3
Las enfermedades pulmonares, en especial la EPOC, también las enfermedades del intersticio y el sindrome de fibrosis pulmonar combinado con enfisema (SCEF), son la causa más frecuente de HP, después de la ECI (Ver Grupo 3 Tabla 4). Suele presentarse con cifras moderadas de presión pulmonar y tiene mal pronóstico5, 10, 186.
La presencia de HP es un factor de riesgo de mortalidad con una correlación inversa entre la PAPm o la RVP, o ambas, y la tasa de supervivencia187, 188. Valores de PAPm mayores a 35 mmHg tienen una prevalencia solo del 3-5%. La proporción de pacientes con fibrosis pulmonar idiopática (FPI) y PAPm > 25 mmHg oscila del 8% a más del 60% en correlación con la gravedad de la enfermedad de base, dependiendo del momento en que son evaluados. Una PAPm >40 mmHg en estadios finales de la enfermedad no es frecuente (9%); no obstante, no existe relación entre la gravedad de la HP y la intensidad de la fibrosis en la TC o el deterioro de la función pulmonar. La tendencia a desarrollar HP alcanza el 30 a 40% en aquellos con SCEF en quienes la gravedad de la HP (PAPm 35-45 mmHg) y la disminucion de la DLCO contrastan con volúmenes pulmonares normales o apenas disminuidos sin obstrucción del flujo aéreo187, 188. La presencia de HP es predictor de riesgo tanto en EPOC, FPI como en el SCEF
6.1 Definición
Si bien la definición de HP en el Grupo 3 es la misma que aplica a cualquier HP, se ha acordado abandonar el término “HP desproporcionada” o “HP fuera de proporción”188. En su lugar se ha introducido, para EPOC, FPI y SCEF una clasificación hemodinámica desarrollada en la Tabla 5110, 188.

6.2 Diagnóstico
En estos pacientes, los síntomas son difícilmente separables de los de la enfermedad respiratoria de base. En la EPOC, la PAPm tiene mejor correlación con la hipoxemia en reposo que con el grado de deterioro que permite investigar la espirometría5. Los signos prominentes en pacientes con enfermedad pulmonar y HP son deterioro de la capacidad de ejercicio, intensificación de la hipoxemia y disminucion de la supervivencia. Los signos más sugestivos de HP en pacientes con enfermedad pulmonar son DLCO desproporcionadamente disminuida con baja PaCO2. En pacientes con HP grave siempre se recomienda descartar ECI y HPTEC10.
El ecocardiograma es la modalidad no invasiva para evaluar la posibilidad de HP en una paciente con EPOC o enfermedad intersticial difusa del parénquima pulmonar188. El CCD no se incluye en la rutina del diagnóstico a excepción de las situaciones puntuales que se enumeran en la Tabla 52188, 189. Las mediciones deben realizarse en condiciones de reposo y con oxígeno suplementario en pacientes que lo requieran. Dado que en el contexto de una enfermedad pulmonar crónica puede desarrollarse una HAP Grupo 1 se han sugerido criterios para el diagnóstico diferencial entre ambas entidades (Tabla 53)188.

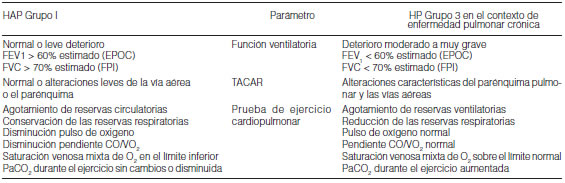
6.3 Tratamiento
El tratamiento se basa en la enfermedad de base y en pacientes con insuficiencia respiratoria se agrega oxigenoterapia crónica domiciliaria que reduce su progresión.
No se recomiendan los vasodilatadores ya que inhiben la vasoconstricción hipóxica y empeoran el intercambio gaseoso. Esto se ha observado por ejemplo con bosentán190, sildenafil191, 192, BCC, y con ON e iloprost inhalado5. Los pacientes con HP grave (PAP > 40mmHg) que no guardan relación con el grado de obstrucción de la vía aérea o hipoxemia deben ser derivados a centros especializados para evaluar la eventual necesidad de tratamientos específicos5, 193.
En la Tabla 54 se describen las modalidades del abordaje diagnóstico terapéutico de HP en pacientes con HP y enfermedad pulmonar.

Ante la ausencia de ensayos clínicos aleatorizados, controlados en pacientes con neumopatía con patrones restrictivos y obstructivos e HP que permita orientar la indicación de terapias específicas se han realizado la siguiente clasificación y recomendaciones terapéuticas. En términos generales se clasifica a los pacientes en categorías. Las categorías 1 y 2 se describen en la Tabla 55188. Pertenecen a la tercera categoría los pacientes con enfermedad pulmonar restrictiva u obstructiva o combinación de ambas, e HP grave según la definición mencionada más arriba; su pronóstico es pobre y se los suele manejar con “tratamientos compasivos” o se los invita a participar en ensayos clínicos. En la cuarta categoría, no incluida en la tabla, quedan los pacientes en fases avanzadas, terminales, con una expectativa de vida limitada. En estos casos se está investigando el uso de ventilación mecánica no invasiva domiciliaria y oxigenación por membrana extracorpórea como pasos para prolongar la supervivencia y la espera hasta un trasplante188.
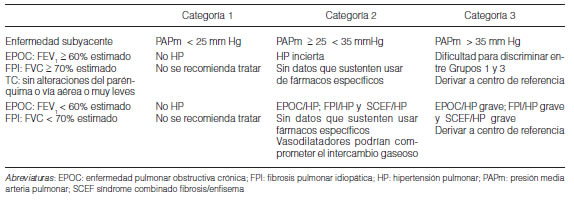
7. Hipertensión pulmonar tromboembólica crónica y otras obstrucciones de la arteria pulmonar - Grupo 4
La hipertensión pulmonar tromboembólica crónica (HPTEC) se produce por una oclusión tromboem-bólica no resuelta de los vasos pulmonares y por remodelamiento vascular. Afecta principalmente a vasos grandes, lo que la hace pasible, en general, de tratamiento quirúrgico.
Si bien no se conocen datos exactos, la incidencia acumulativa de esta enfermedad es del 0.1 al 9.1% 24 meses después del primer evento de tromboembolismo pulmonar agudo194. En Estados Unidos, se han estimado 2500 casos por año. Algunos datos sugieren que esta condición puede ocurrir en, aproximadamente, 5 individuos/1.000.000 habitantes/año195. El 3.8% de los pacientes que sobrevivieron al primer episodio vuelven a embolizar en los dos años siguientes. Si bien se considera un trastorno secundario a tromboembolismo venoso, el 40% de los individuos no tiene ese antecedente clínico y, sólo algunos factores de riesgo tromboembólicos clásicos están ocasionalmente presentes (factor VIII elevado y anticuerpos antifosfolípidos)5, 196, 197. También se lo ha asociado con esplenectomía, derivación ventrículo-atrial por hidrocefalia, osteomielitis crónica y enfermedad inflamatoria intestinal.
7.1 Diagnóstico
La edad promedio de los pacientes en el momento del diagnóstico es de 63 años, con compromiso por igual de ambos sexos. Las manifestaciones clínicas son inicialmente inespecíficas, con disnea de esfuerzo, tos, edema, hemoptisis y evidencias de insuficiencia VD en etapas avanzadas. El intervalo de tiempo entre el inicio de los síntomas y el diagnóstico es de alrededor de 14 meses. El diagnóstico diferencial incluye embolia por células tumorales o cuerpo extraño, sarcoma de la AP, hidatidosis, estenosis congénita de la AP (Ver Tabla 4).
Los estudios para la detección de HPTEC no son una práctica de rutina en caso de enbolia pulmonar, ya que en muchos casos no se constata el antecedente. El centellograma pulmonar de ventilación-perfusión (V/Q) planar es el examen de primera elección para diagnosticar HPTEC en pacientes con HP, con una sensibilidad del 96 a 97%, y especificidad del 90 a 95%5, 10, 86, 194, 198. Un examen normal o con defectos no segmentarios excluye el diagnóstico199. Tanto el centellograma pulmonar de ventilación-perfusión (V/Q) como la angiografía por tomografía computada tienen una eficacia diagnóstica excelente. La angiografía por cateterismo selectivo de la arteria pulmonar (con sustracción digital), en las proyecciones antero-posterior y lateral, es el estándar de oro para el diagnóstico y confirmación de la HPTEC y la evaluación de la operabilidad. Su mayor ventaja es que combina las imágenes con la evaluación de los parámetros hemodinámicas usando CCD. Ambas pruebas son requeridas para confirmar el diagnóstico, localizar los trombos y determinar la gravedad. Estos exámenes son sumamente útiles para definir la indicación y el riesgo quirúrgicos previamente a la endarterectomía. Están indicados en caso de hallarse alteraciones en el centellograma pulmonar V/Q10, 198. Diferenciar la HPTEC de la embolia pulmonar subaguda requiere tres meses de tratamiento anticoagulante, aunque algunos pacientes con obstrucción unilateral completa pueden presentar una hemodinamia normal en reposo aún en presencia de síntomas (Tabla 56).

7.2 Tratamiento
7.2.1 Quirúrgico
La endarterectomía pulmonar bilateral a través de la capa media de la arteria pulmonar, bajo hipotermia profunda y paro circulatorio, sin necesidad de perfusión cerebral, continúa siendo la técnica quirúrgica de elección para tratar la HPTEC. Como regla, esta opción nunca debe ser excluida ya que cuando es técnicamente posible, es una opción con altas probabilidades de curación; la operabilidad debe ser evaluada por un equipo quirúrgico y clínico antes de considerar otras opciones terapéuticas y se recomienda solicitar una segunda opinión en caso que la primera indique no operar5, 10, 198, 200. La mortalidad intrahospitalaria en centros especializados, en Europa, ronda el 4.7%; en centros con alta experiencia en Estados Unidos la mortalidad es inferior201. Un centro ideal debe realizar 20 cirugías anuales con una mortalidad menor al 10%2, 3. Generalmente, es necesaria una evaluación preoperatoria detallada, el acceso a un equipo quirúrgico con experiencia y un adecuado manejo perioperatorio. Los criterios generales de operabilidad incluyen CF OMS II-IV; actualmente se considera que la edad per se no es contraindicación para la cirugía ni tampoco los valores de RVP ni la presencia de disfunción del VD5, 10. (Figura 6). La aplicación de soporte vital extracorpóreo como la oxigenación por membrana extracorpórea es una medida de sostén efectiva posquirúrgica y se recomienda para prevenir complicaciones graves (edema de reperfusión)10, 198. En pacientes no intervenidos y en aquellos con HP persistente la supervivencia es pobre. Después de la intervención se recomienda una evaluación hemodinámica a los seis y doce meses, en un centro de referencia10.
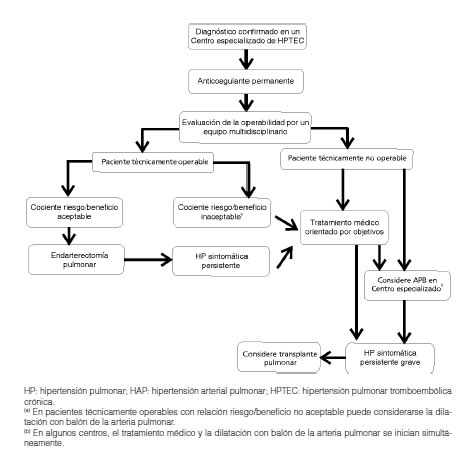
Aunque la dilatación de las arterias pulmonares con balón no tiene un uso extendido, está ganando atención en todo el mundo. Sin embargo, por el momento, su implementación se limita a centros con experiencia y alto volumen de pacientes con HPTEC. Para esta técnica se usan pequeños balones, tratando un sólo lóbulo cada vez, repitiendo las sesiones según necesidad. La intervención es asistida utilizando imágenes intravasculares. La técnica debe ser cuidadosa a fin de evitar edema pulmonar de reperfusión.
7.2.2 Terapia farmacológica
Los pacientes deben permanecer anticoagulados de por vida con un RIN de 2 a 3 para prevenir recurrencias, aún después de la endarterectomía5, 10, 202. El tratamiento medicamentoso especifico para HP no reemplaza a la cirugía, únicamente está indicado en casos técnicamente inoperables o con una relación riesgo/beneficio inaceptable o con HP posendarterectomía (Tabla 57)5, 198.

Se han realizado algunos estudios con prostanoides, AREs e IPDE-5 que sugirieron mejoría hemodinámica y clínica, con mejor supervivencia que los grupos control5, 203. Se necesitan más estudios con los fármacos de esos grupos para evaluar sus beneficios en esta patología. Hasta el momento riociguat, estimulante de la enzima guadenilato ciclasa soluble, es el primer y unico fármaco aprobado en EEUU, Europa y tambien en Argentina, que ha mostrado eficacia en ensayo clínico aleatorizado, controlado para HPTEC inoperable o persistente después de la endarterectomía (Figura 6 y Tabla 58)198, 135.
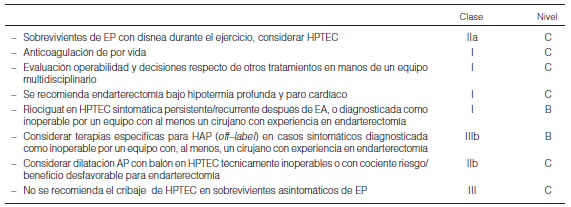
8. Hipertensión pulmonar con mecanismos multifactoriales y/o no esclarecidos - Grupo 5
La HP Grupo 5 incluye numerosas etiopatogenias cuyos mecanismos incluyen vasoconstricción pulmonar, vasculopatía proliferativa, compresión extrínseca, oclusión intrínseca, obliteración vascular e insuficiencia cardiaca izquierda (Ver Tabla 4). El diagnóstico pretende identificar la causa para poder instaurar un tratamiento dirigido. En este contexto el abordaje terapéutico de la HP es secundario y se rige por el siguiente axioma “tratar al pulmón no a la presión”. No hay evidencias aportadas por ensayos clínicos aleatorizados acerca del uso de terapias específicas pero debe tenerse en cuenta que muchas de estas entidades tienen un componente venoso (EVOP) que podría empeorar si se usan vasodilatadores de la arteria pulmonar10.
9. Centro de referencia en hipertensión pulmonar
La complejidad de la HP requiere el abordaje de un equipo multidisciplinario especializado en el tema, con experiencia en diagnóstico, seguimiento y tratamiento. Las sociedades científicas internacionales recomiendan la creación de centros de referencia en hipertensión pulmonar (CRHP) definidos como “una unidad asistencial caracterizada por su competencia específica en la atención de personas con HP, para lo cual cuenta con la infraestructura necesaria y recursos humanos de alta especialización”2, 3, 10, 204, 205. En Estados Unidos y Europa estos centros tienen un caudal de pacientes, entre nuevos y crónicos en tratamiento, de alrededor de 200 por año. Para ser considerado como tal se deben asistir un mínimo de 50 pacientes con HAP o HPTEC por año, y al menos dos nuevas derivaciones por mes con HAP o HPTEC documentada (IIa-C)10. Para hablar de cantidad de pacientes/año necesarios para ser considerado CRHP habría que aclarar ese número a que país se corresponde y la cantidad de habitantes de dicho país. A su vez si ese país tiene patologías prevalentes como la altura en México o la esquistosomiasis en Brasil. En Argentina estimamos apropiada una cantidad de un paciente nuevo por mes y veinte pacientes en seguimiento por año. Para la población pediátrica, tomados los datos aportados por las Guías Europeas, se establece una cifra de 30-50 pacientes/año. Con este caudal se aseguran mejores resultados y, además, se puede desarrollar investigación. Un punto clave a tener en cuenta es que en caso de no disponerse alguna de las prestaciones mencionadas deberá establecerse enlaces con otras instituciones a fin de asegurar la atención adecuada.
La existencia de CRHP no invalida que grupos de trabajo debidamente calificados puedan trabajar en conjunto sin tener una única ubicación geográfica siguiendo las guías de buena práctica clínica para el manejo de la hipertensión pulmonar. Un CRPH optimiza la utilización de recursos, coordina acciones con Niveles inferiores de atención y dispone de datos para incluir en un necesario Registro Nacional.
9.1 Derivación al CRHP
Se recomienda derivar a un CRHP a aquellos los pacientes con sospecha de HAP, HPTEC, HP multifactorial, así como a aquellos con enfermedad respiratoria o cardíaca e HP severa, para confirmar o excluir el diagnóstico. La derivación no debe demorarse, ya que la mayor parte de los pacientes se deteriora rápidamente114, 204, 206.
Criterios de derivación
– Detección de HP por ecocardiograma Doppler con velocidad de regurgitación tricuspídea > 3,4 m/seg.
– Síntomas sospechosos de HP (CF II-IV), enfermedades asociadas o factores de riesgo de HP, aunque la velocidad de regurgitación tricuspídea < 3,4 l/seg.
– Sospecha de HPTEC.
– HP grave asociada a otras patologías (enfermedad respiratoria crónica, ECI).
– Disfunción y dilatación del ventrículo derecho en el ETT.
– Signos y síntomas de insuficiencia cardíaca derecha de etiología incierta.
– Pacientes con HP con tratamiento adecuado que no mejoran o empeoran su evolución.
9.2 Recursos humanos2, 3, 5, 10, 114, 204, 205
– Dos médicos como planta permanente, especialistas en neumonología y cardiología con experiencia en ecocardiograma, CCD y PVRPA, con sólida formación y experiencia en el manejo de la enfermedad, sesiones clínicas dedicadas a HP para pacientes ambulatorios, hospitalizados y encuentros multidisciplinarios.
– Licenciado en enfermería con especial dedicación en el centro, con interés y experiencia en la enfermedad y sus distintos tratamientos.
– Radiólogo con experiencia en interpretación de imágenes de HP
– Licenciado en psicología
– Asistente social
– Un plantel de consultores de otras especialidades: reumatólogos, clínicos, hematólogos, infectólogos, psiquiatras, gastroenterólogos, hepatólogos, radiólogos, anestesistas, hemodinamistas y cirujanos torácicos y cardiovasculares, telefónicamente disponibles.
(I-C)10.
Uno de los profesionales médicos deberá actuar como coordinador del CRHP; la figura de un co-coordinador es aconsejable a los efectos de complementar/reemplazar al coordinador de acuerdo a las circunstancias. Es importante incorporar a la estructura personal administrativo de apoyo, para coordinar la atención de los pacientes.
9.3 Requisitos de servicios permanentes2, 3, 5, 10, 114, 204, 205
– Personal médico en internación con experiencia en HP.
– Unidad de cuidados intensivos con experiencia en el manejo de HP.
– Servicio de consulta externa especializado en pacientes ambulatorios.
– Servicio de emergencias disponible las 24 horas.
– Acceso a medicamentos específicos para HAP y HPTEC acorde a las regulaciones de cada país.
– Base de datos con registro de al menos tres pacientes nuevos/año y un mínimo de 20 pacientes en seguimiento.
9.4 Estudios diagnósticos2, 3, 5, 10, 114, 204, 205
– Laboratorio bioquímico y hematológico e inmunológico (autoanticuerpos) y biomarcadores (PROBNP-NtPROBNP).
– Radiología convencional.
– Ecografía Doppler cardiaca transtorácica y transesofágica, y periférica
– TACAR y angio TACAR.
– Estudios completos de función pulmonar (Espirometría sin y con broncodilatación, Volúmenes pulmonares, DLCO, PM6M, PECP).
– Medicina Nuclear (Centellograma pulmonar de V/Q).
– Laboratorio de sueño.
– Resonancia magnética nuclear.
– Pediatría (referimos al Capítulo de HP en pediatría).
– Servicio de Hemodinamia con experiencia en la realización de CCD y PVRPA. Mínimo 20 PVRPA al año en pacientes con HAPI, HAPH, HAP inducida por fármacos o drogas. (IIa-C)10.
9.5 Red de interconexión con otros servicios con posibilidades de rápida derivación
– Genética
– Planificación familiar
– Trasplante pulmonar
– Otras especialidades (Reumatología, gastroenterología, hematología, infectologia)
– Enfermedades congénitas cardiacas del adulto
– Rehabilitación cardio-respiratoria (I-C)10.
De no disponer la Institución de capacidad para cirugía de tromboendarterectomía pulmonar, trasplante pulmonar o cardio-pulmonar, debería proveer los medios necesarios para una pronta derivación a centros que aporten dichas prestaciones cuando sean requeridas204.
9.6 Actividades académicas
– Participación en ensayos clínicos colaborativos Fase II y III en pacientes con HAP y HPTEC (IIa-C)10.
– Programas de auditoría de adherencia a los lineamientos, resultados clínicos y sobrevida.
– Educación sobre criterios de derivación.
– Participación en la creación de una red de centros de referencia.
– Comunicación con asociaciones de pacientes.
10. Comentarios finales y conclusión
En el presente artículo se comenta el desarrollo de las primeras Guias Argentinas de Consenso de Diagnóstico y Tratamiento de la Hipertensión Pulmonar (HP).
– Esta guía sobre HP establece una nueva clasificación unificada para adultos y niños, modifica la definición hemodinámica de la HP poscapilar y revisa los nuevos conceptos fisiopatológicos de esta entidad.
– Se ofrecen también nuevos algoritmos diagnósticos y terapéuticos, en los que se indica la necesidad de remitir a estos pacientes a centros especializados en su diagnóstico y tratamiento.
– Se modifica también la evaluación de la gravedad de esta entidad, se establecen objetivos terapéuticos y se presentan los nuevos fármacos y las estrategias para combinarlos.
– Se ha clarificado y completado la definición hemodinámica de HP con la incorporación de RVP > 3 UW a la definición de HAP y la introducción de nuevos parámetros hemodinámicos en la HP poscapilar. La combinación de GDTP y RVP permite una mejor tipificación.
– La guía realiza una actualización y ampliación de la clasificación clínica incluyendo entidades propias de la edad pediátrica y mutaciones genéticas.
– El Grupo 1’, EPVO/HCP, se ha ampliado e incluye HAPI, HAPH, HAPD o radiación y formas asociadas.
– Se incorpora la HP en edad pediátrica a la clasificación con especial mención a las cardiopatías congénitas como causa subyacente de HP pre-capilar y post-capilar y la HP asociada a las anomalías del desarrollo pulmonar.
– Los datos epidemiologicos de HAP pertenecen a estudios realizados realizados en otros sitios del planeta y registros realizados en nuestro país.
– Los avances mundiales en genética se han centrado en los pacientes con HAPI o HAPH y la EPVO.
– No se ha encontrado ningún sustrato en los pacientes con formas de HAP combinadas o con HP de los Grupos 2 al 5.
– También se realizan consideraciones relevantes en relación al diagnóstico:
– En las pruebas de función respiratoria destaca el papel del examen de DLCO. Si es < 45%, obliga a estudiar detenidamente enfermedad respiratoria asociada y descartar EPVO. La DLCO disminuida es un marcador de mal pronóstico.
– En el ecocardiograma Doppler se recomienda no utilizar la estimación no invasiva de la PSAP como criterio diagnóstico. Basándose en los hallazgos ecocardiográficos, se distinguen tres Niveles de probabilidad de sufrir HP (bajo, medio y alto) que implican, respectivamente, concluir el estudio, repetirlo al cabo de un periodo determinado o continuarlo. Esta división se establece según la velocidad máxima de regurgitación tricuspídea y la presencia de signos ecocardiográficos indicativos de HP.
– El CCD diagnóstico es la prueba obligada para el diagnóstico de HAP y la recomendable para valorar la respuesta terapéutica.
– Para la PVRPA el ON es el estandar de oro pero se pueden emplear otros darmacos, tal y como se menciona en el texto. Es de destacar que se establecen indicaciones muy precisas para la realización correcta de una técnica, indicando meticulosamente la forma de obtener y garantizar mediciones con la máxima precisión y señalando qué parámetros son imprescindibles y cómo hay que medirlos.
– Se realiza una clasificación según el riesgo de los pacientes en 3 grupos, y se asigna la probabilidad de mortalidad a 1 año: riesgo bajo (< 5%), intermedio (5-10%) y alto (> 10%). El objetivo principal es mantener al paciente en el grupo de bajo riesgo. De hecho, el grupo de riesgo intermedio se considera subóptimo.
– El tratamiento ha sido objeto de un meticuloso abordaje:
– El tratamiento específico de la HAP aporta importantes novedades en esta guía, con la aparición de los nuevos fármacos y especialmente, la incorporación de una escala jerárquica basada en el los criterios primarios de valoración de los ensayos clínicos realizados, con la intención de aunar el Nivel de evidencia y la eficacia clínica.
– Se introduce el novedoso concepto de tratamiento combinado de inicio lo que podria representar un cambio de paradigma en el futuro. Así, los fármacos en monoterapia o en combinación en ensayos clínicos en los que el objetivo primario era el tiempo hasta el deterioro o el evento clínico o la mortalidad por cualquier causa o la respuesta insatisfactoria se destacan en el enfoque terapéutico.
– Se continua con la recomendación de anticoagulación crónica para pacientes con HAP idiopática, heredable y asociada a anorexígenos. La anticoagulación se considera especialmente indicada para los pacientes portadores de catéteres centrales permanentes.
– Se detallaron las características que debe tener un centro para ser de referencia: volumen mínimo de pacientes; personal y tecnología disponibles; metodología de trabajo; comunicación en red con otros hospitales, investigación y docencia.
Aspectos aún no aclarados
– Se establece como valor normal de PAPm 14 mmHg en reposo, con el límite superior de la normalidad en 20 mmHg. Sin embargo, se define la HP como PAPm ≥ 25 mmHg. Los valores intermedios, entre 20 y 24 mmHg, tienen un significado no aclarado y se recomienda un seguimiento estrecho en poblaciones de riesgo, especialmente esclerodermias y familiares de pacientes con HAPH. No se indica una pauta específica.
– No se aporta una definición de HP en ejercicio por falta de información confiable que permita establecer los límites de las respuestas normal y patológica, pese a reconocerse que el comportamiento de la HP en ejercicio puede ser una excelente herramienta para el diagnóstico precoz y la evaluación pronóstica.
– El diagnóstico diferencial entre HAP y HP asociada a insuficiencia cardíaca con fracción de eyección conservada sigue siendo un punto crítico y de difícil decisión en la práctica clínica diaria.
– Queda reflejada en la guía la utilidad del cateterismo con sobrecarga de volumen para pacientes con perfil clínico que apunte a HP del grupo 2 en los que se objetiva una PAPE ≤ 15 mmHg. Sin embargo, se reconoce la falta de estandarización de la prueba y la escasa información disponible sobre su confiabilidad para establecer puntos de corte claros.
– El CCD en ejercicio podría ser una herramienta útil, pero tampoco está protocolizado y la complejidad de su realización lo hace poco aplicable en la práctica.
– Es destacable la indicacion de entrenamiento supervisado de pacientes con HAP. Sin embargo, todavía se desconoce cuál es el mejor método de entrenamiento y el impacto real en el pronóstico a largo plazo.
– La anticoncepción es fundamental. No obstante, no hay acuerdo sobre cuál es el mejor método anticonceptivo. Se recomienda combinar un método de barrera con anovulatorios (compuestos por progestágenos únicamente). En una paciente con HAP, la colocación de un dispositivo intrauterino liberador de levonorgestrel requiere acceso a un equipo de anestesia entrenado en el manejo de una posible reacción vasovagal inducida por la manipulación del cuello del útero.
– La mayor parte de los trasplantes son bipulmonares y solo se indica trasplante cardiopulmonar en los casos de disfunción irreversible del VD o el ventrículo izquierdo o para pacientes con cardiopatías congénitas complejas. Sin embargo, siguen sin establecerse unos límites claros que indiquen cuándo la disfunción ventricular es irreversible y hay que realizar un trasplante cardiopulmonar.
11. Resumen de aspectos destacados
En la definición hemodinámica de HAP, se completa la PAPm > 25 mmHg con la exigencia de RVP > 3 UW.
Se introduce una nueva definición hemodinámica para la HP combinada precapilar y poscapilar: GDTP ≥ 7 mmHg y RVP > 3 UW.
Se incorpora a la clasificación entidades propias de la edad pediátrica que afectan a los cinco grupos de HP.
Se aclara fehacientemente el listado de los factores de riesgo de HP.
Se recomienda el cribado anual de los pacientes con esclerodermia (ecocardiograma, DLCO y BNP) asintomáticos (I-C).
Se destaca el valor de la DLCO, que debe realizarse siempre en el momento del diagnóstico (I-C).
En el ecocardiograma se distinguen tres Niveles de probabilidad de sufrir HP (bajo, medio y alto) según la velocidad máxima de regurgitación tricuspídea y la presencia de “signos ecocardiográficos de HP”.
Las valoraciones general del ecocardiograma y del riesgo de HP determinarán la indicación de realizar CCD.
El CCD es imprescindible para el diagnóstico de HAP y HPTEC (I-C).
Se especifica el procedimiento para realizar el CCD y el test vasodilatador y se recomienda realizarlo en centro experto (I-C).
La PVRpa se recomienda en la HAPI, HAPH y HAPD (I-C).
Se realiza una clasificación según el riesgo de los pacientes en 3 grupos asignando una probabilidad de mortalidad a 1 año: riesgo bajo (< 5%), intermedio (5-10%) y alto (> 10%).
Se recomienda la valoración multifactorial de elementos clínicos, bioquímicos, capacidad funcional, ecocardiográficos y hemodinámicos (I-C) a intervalos regulares.
Se considera a los pacientes bien controlados cuando tienen un perfil de riesgo bajo (I-C). Los anticoagulantes están indicados en la HAPI, la HAPH y la HAPD (IIb).
Los BCC están indicados en la HAPI, la HAPH y la HPD con respuesta positiva en la PVDPA, y se debe revaluar hemodinámicamente a los 3-6 meses (I-C).
Recomendación en CF II y III de iniciar tratamiento combinado de entrada o monoterapia. Recomendación en CF-IV de iniciar con tratamiento combinado de entrada que incluya epoprostenol intravenoso (IIa).
El trasplante pulmonar se debe indicar precozmente ante el fallo del tratamiento (I-C). Se debe referir para trasplante pulmonar al paciente con EVOP al diagnóstico.
Es necesario implementar una evaluación pronóstica y un algoritmo terapéutico específico en pediatría (I-C).
Se establecen unos límites hemodinámicos para reparar los cortocircuitos sistémico-pulmonares en los pacientes con HP-CC.
Para los pacientes con HP de los Grupos 2 y 3, el tratamiento con fármacos específicos para la HAP no está indicado (III-C).
Todo paciente con HPTEC deber ser valorado en un centro experto (con cirujano especializado en endarterectomía) para decidir sobre la factibilidad de la cirugía antes de iniciar otros tratamientos.
El riociguat está indicado en la HPTEC no quirúrgica o con HP persistente tras la cirugía (I-B).
Agradecimiento
A Marta Pérez Sainz y a la Dra. Marcela Prado por su por su colaboración en la coordinación, asistencia técnica y preparación de este documento.
Nota
Estas Guías Argentinas de Consenso en Diagnóstico y Tratamiento de la Hipertensión Pulmonar han sido publicadas previamente en la Revista Argentina de Cardiología, vol 85 Supl 3, octubre 2017.
1. McLaughlin V, Archer S, Badesch D, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009; 53: 1573-1619.
2. Galiè N, Hoeper M, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30: 2493-2537.
3. Galie N, Hoeper M, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Respir J 2009; 34: 1219-1263.
4. Galiè N, Hoeper M, Humbert M, et al. Guía de práctica clínica para el diagnóstico y tratamiento de la hipertensión pulmonar. Rev Esp Cardiol 2009; 62: 1464. e1- e58.
5. Sociedad Argentina de Cardiología - SAC; Asociación Argentina de Medicina Respiratoria - AAMR; Sociedad Argentina de Reumatología - SAR. Consenso para el diagnóstico y tratameinto de la Hipertension arterial pulmonar. Revista Argentina de Cardiología 2011; 79 (Supl2).
6. Badesch D, Champion H, Sanchez M, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54: S55-S66.
7. Mazzei J, Caneva J, Perrone S, et al. Actualizaciòn en el diagnóstico y tatamiento de la Hipertensión Pulmonar. Revista Medicina 2011; 71: Supl.1
8. Simonneau G, Gatzoulis M, Adatia I, et al. Updated Clinical Classification of Pulmonary Hypertension. JACC 2013; 62(25, Suppl D): D34-41.
9. Hoeper M, Harm Jan Bogaard, Robin Condliffe, et al. Definitions and Diagnosis of Pulmonary Hypertension J Am Coll Cardiol 2013; 62:D42-50
10. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). European Heart Journa, 2015; doi: 10.1093/eurheartj/ehv317.
11. Herve P, Lau EM, Sitbon O, et al. Criteria for diagnosis of exercise pulmonary hypertension. Eur Respir J 2015; 46(3): 728-737.
12. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006; 173: 1023-1030.
13. Appelbaum L, Yigla M, Bendayan D, et al. Primary pulmonary hypertension in Israel: a national survey. Chest 2001; 119(6): 1801-1806.
14. Hyduk A, Croft JB, Ayala C, Zheng K, Zheng Z, Mensah G. Pulmonary hypertension surveillance--United States, 1980-2002. MMWR Surveill Summ 2005; 54(5): 1-28.
15. Peacock A, Murphy N, McMurray J, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007 Jul; 30(1): 104-109.
16. Deng Z, Morse J, Slager S, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67(3): 737-744.
17. Morse J. Genetic studies of pulmonary arterial hypertension. Lupus 2003; 12(3): 209-212. Review.
18. Nichols W, Koller D, Slovis B, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet 1997; 15(3): 277-280.
19. D’Alonzo G, Barst R, Ayres S, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991; 115: 343-349.
20. Escribano-Subias P, Blanco I, Lopez-Meseguer M, Lopez-Guarch CJ, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J 2012; 40: 596-603.
21. Pepke-Zaba J, Delcroix M, Lang I, et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation 2011; 124: 1973-198.
22. Alves L; Gavilanes F; Jardim C, et al. Pulmonary Arterial Hypertension in the Southern Hemisphere Results From a Registry of Incident Brazilian Cases. Chest 2015; 147(2): 495-501.
23. Shapiro B, McGoon M, Redfield M. Unexplained pulmonary hypertension in elderly patients. Chest 2007; 131(1): 94-100.
24. Yang X, Mardekian J, Sanders K, Mychaskiw MA, Thomas J 3rd. Prevalence of pulmonary arterial hypertension in patients with connective tissue diseases: a systematic review of the literature. Clin Rheumatol 2013; 32(10): 1519-1531.
25. Magliano M, Isenberg D, Hillson J. Pulmonary hypertension in autoimmune rheumatic diseases: where are we now? Arthritis Rheum. 2002; 46(8): 1997-2009.
26. McLaughlin V, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002; 106: 1477-1482.
27. Lee S, Rubin L. Current Treatment strategies for pulmonary arterial hypertension. J.Intern Med 2005; 258: 199-215.
28. Castro O. Systemic fat embolism and pulmonary hypertension in sickle cell disease. Hematol Oncol Clin North Am 1996; 10(6):1289-1303
29. Krowka M, Swanson K, Frantz P, McGoon M, Wiesner R. Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology 2006; 44: 1502-1510.
30. Feltracco P, Serra E, Brezzi ML, et al. Hemodynamic profile of portopulmonary hypertension. Transplant Proc 2009; 41(4): 1235-1239.
31. Speich R, Jenni R, Opravil M, Pfab M, Russi E. Primary pulmonary hypertension in HIV infection. Chest 1991;100(5): 1268-71.
32. Sitbon O, Lascoux-Combe C, Delfraissy J, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J RespirCrit Care Med 2008; 177: 108-113.
33. Morse J, Barst R, Itescu S, Flaster E, Sinha G, Zhang Y, Fotino M. Primary pulmonary hypertension in HIV infection: an outcome determined by particular HLA class II alleles. Am J Respir Crit Care Med 1996; 153(4 Pt 1):1299-1301.
34. Zuber J, Calmy A, Evison J, et al; Swiss HIV Cohort Study Group. Pulmonary arterial hypertension related to HIV infection: improved hemodynamics and survival associated with antiretroviral therapy. Clin Infect Dis 2004; 38(8): 1178-1185.
35. Speich R, Jenni R, Opravil M, Jaccard R. Regression of HIV-associated pulmonary arterial hypertension and long-term survival during antiretroviral therapy. Swiss Med Wkly 2001; 131(45-46): 663-665.
36. Galiè N, Manes A, Palazzini M, et al. Management of pulmonary arterial hypertension associated with congenital systemic to pulmonary shunts and Eisenmenger’s syndrome. Drugs 2008; 68: 1049-1066.
37. Duffels M, Engelfriet P, Berger R, et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry.Int J Cardiol 2007; 120(2): 198-204.
38. Melero Marcelo. Estado actual de la hipertensión arterial pulmonar. Insuficiencia Cardiaca 2009; 4(1): 23-26.
39. Talavera M, Cáneva J; Favaloro L., et al. Hipertensión arterial pulmonar. Registro de un centro de referencia en Argentina. Revista Americana de Medicina Respiratoria 2014; 14(2): 144-152.
40. Mazzei J; Cáneva J, Khoury M, Melero M. Mortalidad por hipertensión arterial pulmonar idiopática en la Argentina. Insuficiencia Cardiaca 2015; 10(3): 111-118.
41. Wilkins M, Gibbs J, Shovlin C. A gene for primary pulmonary hypertension. Lancet. 2000; 356(9237): 1207-1208.
42. Newman J, Fanburg B, Archer S, et al. National Heart, Lung and Blood Institute/Office of Rare Diseases. Pulmonary arterial hypertension: future directions: report of a National Heart, Lung and Blood Institute/Office of Rare Diseases workshop. Circulation 2004; 109(24): 2947-2952.
43. West J, Fagan K, Steudel W, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 2004 Apr 30;94(8): 1109-1114.
44. Sztrymf B, Yaïci A, Girerd B, Humbert M. Genes and pulmonary arterial hypertension. Respiration 2007; 74(2): 123-132
45. Sztrymf B, Yaïci A, Jaïs X, Simonneau G, Humbert M. Idiopathic pulmonary hypertension: what did we learn from genes? Sarcoidosis Vasc Diffuse Lung Dis 2005; 22 Suppl 1: S91-100.
46. Trembath R, Thomson J, Machado R, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345(5): 325-3.
47. Chaouat A, Coulet F, Favre C, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension.Thorax 2004; 59(5): 446-448.
48. Nunes H, Humbert M, Sitbon O, et al. Prognostic factors for survival in human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2003; 167(10): 1433-1439.
49. Ahearn GS, Tapson VF, Rebeiz A, Greenfield JC, Jr. Electrocardiography to define clinical status in primary pulmonary hypertension and pulmonary arterial hypertension secondary to collagen vascular disease. Chest 2002; 122: 524-527.
50. Rich S, Dantzker D, Ayres S, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987; 107: 216-223.
51. Chu J, Kao P, Faul J, Doyle R. High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension. Chest 2002; 122: 1668-73.
52. Fedullo P, Auger W, Kerr K, Rubin L. Chronic thromboembolic pulmonary hypertension N Engl J Med 2001; 345: 1465-1472.
53. Lang I, Madani M. Update on Chronic Thromboembolic Pulmonary Hypertension. Circulation 2014; 130: 508-518.
54. Casserly B., Klinger J. Brain natriuretic peptide in pulmonary arterial hypertension: biomarker and potential therapeutic agent. Drug Des Devel Ther 2009; 3: 269-287.
55. Rajpal S; Hilbun J, Dwary A, Smith T, Mina G, Reddy P, Akkus N. Troponin elevation correlates with pulmonary hypertension and hemolytic burden in sickle cell pain crisis. Eur Heart J 2013: 297. DOI: http://dx.doi.org/10.1093/eurheartj/eht307
56. Leuchte H, Baumgartner R, Nounou M, Vogeser M, Neurohr C, Trautnitz M, Behr J.. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med 2006; 173: 744-750.
57. Leuchte H, El Nounou M, Tuerpe J, et al. N-terminal pro-brain natriuretic peptide and renal insufficiency as predictors of mortality in pulmonary hypertension. Chest 2007; 131: 402-409.
58. Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006; 129: 1313-1321.
59. Blyth K, Groenning B, Mark P, et al. NT-proBNP can be used to detect right ventricular systolic dysfunction in pulmonary hypertension. Eur Respir J 2007; 29: 737-744.
60. Vonk Noordegraaf A, Galiè N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev 2011; 20(122): 243-253.
61. Torbicki A, Kurzyna M, Kuca P, et al. Detectable Serum Cardiac Troponin T as a Marker of Poor Prognosis Among Patients With Chronic Precapillary Pulmonary Hypertension. Circulation 2003; 108: 844-848.
62. Sánchez Román J, Castillo Palma M, García Hernández F, González Leon R. Marcadores biológicos. Utilidad para el control del paciente con hipertensión pulmonar. Arch Bronconeumol. 2011; 47(Supl 7): 21-25.
63. Sciomer S, Magri D, Badagliacca R. Non-invasive assessment of pulmonary hypertension: Doppler-echocardiography. Pulm Pharmacol Ther 2007; 20: 135-140.
64. Batal O, Dardari Z, Costabile C, Gorcsan J, Arena V, Mathier M. Prognostic Value of Pericardial Effusion on Serial Echocardiograms in Pulmonary Arterial Hypertension. Echocardiography 2015; 32(10): 1471-1476.
65. Forfia P, Fisher M, Mathai S, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006; 1;174(9): 1034-1041.
66. Tei C, Dujardin K, Hodge D, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr 1996; 9: 838-847.
67. Bossone E, Bodini BD, Mazza A, Allegra L. Pulmonary arterial hypertension: the key role of echocardiography. Chest 2005; 127: 1836-1843.
68. Bossone E, D’Andrea A, D’Alto M, et al. Echocardiography in Pulmonar Arterial Hypertension: from diagnosis to prognosis J Am Soc Echocardiogr 2013; 26: 1-14.
69. Sun X, Hansen J, Oudiz R, Wasserman K. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol 2003; 41: 1028-1035.
70. Meyer F, Ewert R, Hoeper M, et al. Peripheral airway obstruction in primary pulmonary hypertension. Thorax 2002; 57: 473-476.
71. Miyamoto S, Nagaya N, Satoh T, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 2000; 161: 487-492.
72. Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002; 40: 780-788.
73. Sun X, Hansen J, Oudiz R, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001; 104: 429-435.
74. Fuster V, Sanz J. Pulmonary hypertension: new insights fromtechniques imaging. Rev Esp Cardiol 2007; 60 Suppl 3: 2-9.
75. Ghofrani H, Wilkins M, Rich S. Uncertainties in the diagnosis and treatment of pulmonary arterial hypertension. Circulation 2008; 118: 1195-1201.
76. Coulden R. State-of-the-art imaging techniques in chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc 2006;3: 577-583.
77. Barst R, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 40S-47S
78. Bajc M, Olsson C, Palmer J, Jonson B. In: Freeman LM, editor. Quantitative ventilation/perfusion SPECT (QV/PSPECT): a primary method for diagnosis of pulmonary embolism. Philadelphia: 2004; 2010: 173-186.
79. Tunariu N, Gibbs S, Win Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med 2007; 48: 680-684.
80. Fedullo P, Adams A. Postoperative management of the patient undergoing pulmonary endarterectomy. Semin Thorac Cardiovasc Surg 2006; 18(3): 250-256.
81. Jardim C, Rochitte C, Humbert M, et al. Pulmonary artery distensibility in pulmonary arterial hypertension: an MRI pilot study. Eur Respir J 2007; 29(3): 476-481.
82. Karamitsos T, Francis J, Myerson S, Selvanayagam J, Neubauer S. The role of cardiovascular magnetic resonance imaging in heart failure. J Am Coll Cardiol 2009; 54: 1407-1424.
83. Ley S, Kauczor H, Heussel C, Kramm T, Mayer E, Thelen M, Kreitner K. Value of contrast-enhanced MR angiography and helical CT angiography in chronic thromboembolic pulmonary hypertension. Eur Radiol 2003; 13(10): 2365-2371.
84. Albrecht T, Blomley M, Cosgrove D, et al. Non-invasive diagnosis of hepatic cirrhosis by transit-time analysis of an ultrasound contrast agent. Lancet 1999; 353:1579-1583.
85. Friedrich-Rust M, Rosenberg W, Herrmann E, Zeuzem S, Sarrazin C. Comparison of ELF, FibroTest and FibroScan for the non-invasive assessment of liver fibrosis. BMC Gastroenterol 2010; 10: 103.
86. Hoeper M, Lee SH, Voswinckel R, Palazzini M, Jais X, Marinelli A, et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol 2006; 48: 2546-2552.
87. Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105-3111.
88. Hoeper M, Maier R, Tongers J, et el. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am J Respir Crit Care Med 1999; 160 ;535-541
89. Bush C, Stang J, Wooley C, Kilman J. Occult constrictive pericardial disease. Diagnosis by rapid volume expansion and correction by pericardiectomy. Circulation 1977; 56: 924-930.
90. Newman y col Association of the Metabolic Syndrome With Pulmonary Venous Hypertension Chest 2009; 136(1): 31-36.
91. Vachiéry J, Adir Y, Barberá J, et al. y col. Pulmonary Hypertension Due to Left Heart Disease. JACC 2013; 62(25, Suppl D): 100-108.
92. Austin E, Loyd J. The Genetics of Pulmonary Arterial Hypertension. Circ Res 2014; 115: 189-200.
93. Tron V, Magee F, Wright J, Colby T, Churg A. Pulmonary capillary hemangiomatosis. Hum Pathol 1986; 17: 1144-1150.
94. Burke A, Virmani R. Evaluation of pulmonary hypertension in biopsies of the lung. Diagnostic Histopathology 1996; 3(1): 14-26.
95. Kawut S, Taichman D, Archer-Chicko C, Palevsky H, Kimmel S. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest 2003; 123: 344-350.
96. Mandel J, Mark E, Hales C. Pulmonary veno-occlusive disease. Am J Respir Crit Care Med 2000; 162: 1964-73.
97. Humbert M, Sitbon O, Yaici A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J 2010; 36: 549-555.
98. Thenappan T, Shah S, Rich S, Tian L, Archer S, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J 2010; 35: 1079-1087.
99. Benza R, Miller D, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension. Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010; 122: 164-172.
100. Kuhn K, Byrne D, Arbogast P, Doyle T, Loyd J, Robbins I. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med 2003; 167: 580-586.
101. Raymond R, Hinderliter A, Willis P, et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol 2002; 39: 1214-1219.
102. Mahapatra S, Nishimura R, Oh J, McGoon M. The prognostic value of pulmonary vascular capacitance determined by Doppler echocardiography in patients with pulmonary arterial hypertension. J Am Soc Echocardiogr 2006; 19: 1045-1050.
103. Courand P, Jomir G, Khouatra C, et al. Prognostic value of right ventricular ejection fraction in pulmonary arterial hypertension. Eur Respir J 2015; 45(1): 139-149.
104. Wensel R, Opitz C, Anker S, et al. Assessment of survival in patients with primary pulmonary hypertension: importance of cardiopulmonary exercise testing. Circulation 2002; 106 :319-324.
105. Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients withpulmonary hypertension.Chest 2006; 129(5): 1313-1321.
106. Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation 2000; 102: 865-870.
107. Park M, Scott R, Uber P, Ventura H, Mehra M. Usefulness of B-type natriuretic peptide as a predictor of treatment outcome in pulmonary arterial hypertension. Congest Heart Fail 2004; 10: 221-225.
108. Benza R, Gomberg-Maitland M, Miller D, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012; 141(2): 354-362.
109. Mereles D, Ehlken N, Kreuscher S, et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 2006; 114: 1482-1489.
110. Weinstein A, Chin L, Keyser R, et al. Effect of aerobic exercise training on fatigue and physical activity in patients with pulmonary arterial hypertension. Respir Med 2013; 107: 778-784.
111. Chan L, Chin LM, Kennedy M, et al. Benefits of intensive treadmill exercise training on cardiorespiratory function and quality of life in patients with pulmonary hypertension. Chest 2013; 143: 333-343.
112. National Pulmonary Hypertension Centres of the UK and Ireland. Consensus statement on the management of pulmonary hypertension in clinical practice in the UK and Ireland. Thorax 2008; 63: ii1-ii41.
113. de Man F, Handoko M, Groepenhoff H, et al. Effects of exercise training in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2009; 34: 669-675.
114. Barberà J, Escribano P, Morales P, et al. Standards of care in pulmonary hypertension. Consensus Statement of the Spanish Society of Pulmonology and Thoracic Surgery (SEPAR) and the Spanish Society of Cardiology (SEC). Arch Bronconeumol 2008; 44: 87-99.
115. Jaïs X, Olsson K, Barbera J, et al. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur Respir J 2012; 40: 881-885.
116. Gordon C, Collard C, Pan W. Intraoperative management of pulmonary hypertension and associated right heart failure. Curr Opin Anaesthesiol 2010; 23: 49-56.
117. Recommended adult immunization schedule: United States, October 2007-Setember 2008. Ann Intern Med 2007; 147: 725-729.
118. Shafazand S, Goldstein MK, Doyle R, et al. Health-related quality of life in patients with pulmonary arterial hypertension. Chest 2004; 126: 1452-1459.
119. Hopkins R, Morton J, Glissmeyer E, et al. Cognitive dysfunction in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2003; 167:A273.
120. Fuster V, Steele P, Edwards W, Gersh B, McGoon M, Frye R. Primary pulmonary hypertension: natural history and the importance of thrombosis Circulation 1984; 70: 580-587.
121. Rich S, Kaufmann E, Levy P. The effect of high doses of calciumchannel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327: 76-81.
122. Galiè N, Torbicki A, Barst R, et al; Task Force. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 2004; 25: 2243-2278.
123. Roberts D, Lepore J, Maroo A, Semigran M, Ginns L. Oxygen therapy improves cardiaca index and pulmonary vascular resistance in patients with pulmonary hypertension. Chest 2001; 120: 1547-1555.
124. Ghofrani H, Voswinckel R, Reichenberger F, et al. Hypoxia- and non-hypoxia-related pulmonary hypertension - Established and new therapies. Cardiovascular Research 2006; 72: 30-40.
125. Galiè N ; Corris P; Frost A., et al. Updated Treatment Algorithm of Pulmonary Arterial Hypertension. J Am Coll Cardiol 2013; 62: D60-72.
126. British Thoracic Society Standards of Care Committee. Managing passengers with respiratory disease planning air travel: British Thoracic Society recommendations. Thorax 2002; 57: 289-304.
127. Galie N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res 2004; 61: 227-237.
128. Galie´ N, Olschewski H, Oudiz R, et al. Ambrisentan for the treatment of pulmonary arterial hypertension. Results of the ambrisentan in pulmonary arterial hypertension, randomized, doubleblind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008; 117: 3010-3019.
129. Humbert M, Barst R, Robbins I, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J 2004; 24: 353-359.
130. Galiè` N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomized controlled trial. Lancet 2008; 371: 2093-2100.
131. Galie N, Beghetti M, Gatzoulis M, et al. Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter,double-blind, randomized, placebo-controlled study. Circulation 2006; 114: 48-54.
132. Pulido T, Adzerikho I, Channick R, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013; 369(9): 809-818.
133. Galie N, Brundage B, Ghofrani HA, et al. Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119: 2894-2903.
134. Jing Z, Yu Z, Shen J, et al. Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med 2011; 183: 1723-1729.
135. Ghofrani H, Galie` N, Grimminger F, et al. for the PATENT-1 Study Group. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013; 369: 330-340.
136. Galie N, Humbert M, Vachiery J, et al. Arterial Pulmonary Hypertension and Beraprost European (ALPHABET) Study Group. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomised, double-blind placebocontrolled trial. J Am Coll Cardiol 2002; 39: 1496-1502.
137. Badesch D, Tapson V, McGoon et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial [see comments]. Ann Intern Med 2000; 132: 425-434.
138. Olschewski H, Simonneau G, Galie N, et al. for the Aerosolized Iloprost Randomized Study Group. Inhaled iloprost in severe pulmonary hypertension. N Engl J Med 2002; 347: 322-329.
139. Simonneau G, Barst R, Galiè N, et al. Treprostinil Study Group. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension. A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165: 800-804.
140. McLaughlin V, Rubin L, Benza R, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010; 55: 1915-1922.
141. Hiremath J, Thanikachalam S, Parikh K, et al. TRUST Study Group. Exercise improvement and plasma biomarker changes wiht intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant 2010; 29: 137-149.
142. Jing Z, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation 2013; 127: 624-633.
143. Sitbon O, Channik R, Chin K, et al. for the GRIPHON Investigators. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015; 373: 2522-2533.
144. Galie N, Palazzini M, Manes A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J 2010; 31: 2080-2086.
145. Kemp K, Savale L, O’Callaghan D, et al. Usefulness of first-line combination therapy with epoprostenol and bosentán in pulmonary arterial hypertension: an observational study. J Heart Lung Transplant 2012; 31: 150-158.
146. Galie N, Barbera JA, Frost A, et al. Usefulness of first-line combination therapy with epoprostenol and bosentán in pulmonary arteri New Engl J Med 2015; 379(9): 834-844.
147. Fuehner T, Kuehn C, Hadem J, et al. Extracorporeal membrane oxygenation in awake patients as bridge to lung transplantation. Am J Respir Crit Care Med 2012; 185: 763-768.
148. Keogh A, Mayer E, Benza R, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol 2009; 54: S67-S77
149. Sandoval J, Gaspar J, Peñas H, et al. Effects of atrial septostomy on the survival of patients with severe pulmonary arterial hypertension. Eur Respir J 2011; 38: 1343-1348.
150. Christie J, Edwards L, Kucheryavaya A, et al. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report - 2012. J Heart Lung Transplant 2012; 31: 1073-1086.
151. Weill D, Benden C, Corris P et al. A consensus document for the selection of lung transplant candidates. J Heart Lung Transplant 2015; 34: 1-15.
152. Toyoda Y, Thacker J, Santos R, et al. Long-term outcome of lung and heart-lung transplantation for idiopathic pulmonary arterial hypertension. Ann Thorac Surg 2008; 86: 1116-1122.
153. Fadel E, Mercier O, Mussot S, et al. Long-term outcome of doublelung and heart-lung transplantation for pulmonary hypertension: a comparative retrospective study of 219 patients. Eur J Cardiothorac Surg 2010; 38: 277-284.
154. de Perrot M, Granton J, McRae K, et al. Outcome of patients with pulmonary arterial hypertension referred for lung transplantation: a 14- year single-center experience. J Thorac Cardiovasc Surg 2012; 143: 910-918.
155. INCUCAI 2010. Normativas de Trasplante Intratorácico. En: http://www.incucai.gov.ar/index.php/institucional/legislacion#normativas-de-trasplante-intratorácico.
156. Lamont E, Christakis N. Prognostic disclosure to patients with cancer near the end of life. Ann Intern Med 2001; 134: 1096-1105.
157. Pellegrino E. Some things ought never be done: moral absolutes in clinical ethics. Theor Med Bioeth 2005; 26: 469-486.
158. Ivy D, Abman D, Barst R, et al. Pediatric Pulmonary Hypertension. JACC 2013; 62(25 Suppl D): 117-126.
159. Mouratian D, Capelli H. Cardiopatías Congénitas del Adulto. PROSAC 2009; Módulo 4 Fasciculo 2: 1851.
160. Barst R, McGoon M, Elliott C, Foreman A, Miller D, IvyD. Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012; 125(1): 113-122.
161. Haworth S, Hislop A.Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001-2006. Heart. 2009; 95(4): 312-317.
162. Lammers A, Adatia I, Cerro MJ, et al. Functional classification of pulmonary hypertension in children: Report from the PVRI pediatric taskforce, Panama 2011. Pulm Circ 2011; 1(2): 280-285.
163. Moledina S, Hislop A, Foster H, Schulze-Neick I, Haworth S. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart 2010; 96(17): 1401-1406.
164. Barst R, Maislin G, Fishman A. Vasodilator therapy for primary pulmonary hypertension in children. Circulation 1999; 99(9): 1197-1208.
165. Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52: 3792-3800.
166. Steen V, Medsger T Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum 2003; 48: 516-522.
167. Chang B, Schachna L, White B, Wigley F, Wise R. Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma. J Rheumatol 2006;33: 269-274.
168. Valerio C, Schreiber B, Handler C, Denton C, Coghlan J. Borderline mean pulmonary artery pressure in patients with systemic sclerosis: transpulmonary gradient predicts risk of developing pulmonary hypertension. Arthritis Rheum 2013; 65: 1074-1084.
169. Shah A, Wigley F. Often forgotten manifestations of Systemic sclerosis. Rheum Dis Clin North Am 2008; 34(1): 221-238.
170. Dorfmuller P, Humbert M, Perros F, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol 2007; 38: 893-902.
171. Sitbon O, Yaïci A, Cottin V, et al. The changing picture of patients with pulmonary arterial hypertension in France. Eur Heart J 2011; 32 Suppl 1: 675-676.
172. Degano B, Yaïci A, Le Pavec J, et al. Long-term effects of bosentan in patients with HIV-associated pulmonary arterial hypertension. Eur Respir J 2009; 33: 92-98.
173. Barbaro G, Lucchini A, Pellicelli A, Grisorio B, Giancaspro G, Barbarini G. Highly active antiretroviral therapy compared with HAART and bosentan in combination in patients with HIV-associated pulmonary hypertension. Heart 2006; 92: 1164- 1166.
174. Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med 2008; 178: 6337-6343.
175. Hoeper M, Krowka M, Strassburg C. Portopulmonary hipertensión and hepatopulmonary syndrome. Lancet 2004; 363: 1461-1468.
176. Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130: 120-126.
177. Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J 2007; 30: 1096-1102.
178. Montani D, Price L, Dorfmuller P, et. al. Pulmonary veno-occlusive disease. Eur Respir J 2009; 33: 189-200.
179. Pietra G, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 2004; 43: 25S-32S.
180. Montani D, O’Callaghan D, Savale L, et al. Pulmonary veno-occlusive disease: recent progress and current challenges. Respir Med 2010; 104: S23-S32.
181. Oudiz R. Pulmonary hypertension associated with left-sided heart disease. Clin Chest Med 2007; 28: 233-241.
182. Dickstein K, Cohen-Solal A, Filippatos G, et al. Task Force for Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of European Society of Cardiology,. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J 2008; 29: 2388-2442.
184. Silke B. Haemodynamic impact of diuretic therapy in chronic heart failure. Cardiology 1994; 84 Suppl 2: 115-123.
185. Lewis G, Shah R, Shahzad K, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation 2007; 116: 1555-1562.
186. Cottin V, Le Pavec J, Prévot G, et al. GERM”O”P. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J 2010; 35: 105-111.
187. Andersen K, Iversen M, Kjaergaard J, et al. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J HeartLung Transplant 2012; 31: 373-380.
188. Seeger W, Adir Y, Barberà J. Pulmonary Hypertension in Chronic Lung Diseases JAmColl Cardiol 2013; 62: D109-116.
189. Nathan S, Cottin V. Chapter 12. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Eur Respir Monogr 2012; 57: 148-60.
190. Stolz D, Rasch H, Linka A, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J 2008; 32: 619- 628.
191. Blanco I, Gimeno E, Munoz PA, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med 2010; 181: 270-278.
192. Collard H, Anstrom K, Schwarz M, Zisman D. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest 2007; 131: 897-899.
193. Thabut G, Dauriat G, Stern J, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest 2005; 127: 1531-1536.
194. Lang IM, Pesavento R, Bonderman D, Yuan JX. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J 2013; 41(2): 462-468.
195. Pepke-Zaba J, Jansa P, Kim N, Naeije R, Simonneau G. Chronic thromboembolic pulmonary hypertension: role of medical therapy. Eur respir J 2013; 41: 985-990.
196. Bonderman D, Turecek P, Jakowitsch J, Weltermann et al. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost 2003; 90: 372-376.
197. Wolf M, Boyer-Neumann C, Parent F, Eschwege V, Jaillet H, Meyer D, Simonneau G. Thrombotic risk factors in pulmonary hypertension. Eur Respir J 2000; 15: 395-399.
198. Kim N, Delcroix M, Jenkins P. Chronic Thromboembolic Pulmonary Hypertension. J Am Coll Cardiol 2013; 62: D92-99.
199. Hoeper M, Barbera J, Channick R, et al. Diagnosis, assessment, and treatment of nonpulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol 2009; 54: S85-S96.
200. Thistlethwaite P, Kaneko K, Madani M, Jamieson S. Technique and outcomes of pulmonary endarterectomy surgery. Ann Thorac Cardiovasc Surg 2008; 14:274-282.
201. Madani M, Auger W, Pretorius V, et al. Pulmonary endarterectomy: recent changes in a single institution’s experience of more than 2,700 patients. Ann Thorac Surg 2012; 94: 97-103.
202. Rubin L, Hoeper M, Klepetko W, Galiè N, Lang I, Simonneau G. Current and future management of chronic thromboembolic pulmonary hypertension: from diagnosis to treatment responses. Proc Am Thorac Soc 2006; 3: 601-607.
203. Jaïs X, D’Armini A, Jansa P, et al; Bosentan Effects in iNopErable Forms of chronic Thromboembolic pulmonary hypertension Study Group. Bosentan for treatment of inoperable chronicthromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol 2008; 52: 2127-2134.
204. Diez A, Naval N, Svetliza G, MazzeiJ, Cáneva J. Centros de referencia en hipertensión Pulmonar Documento elaborado por miembros de la sección circulación pulmonar de la AAMR. Revista Americana de Medicina Respiratoria 2013; 13(4): 207-211.
205. Escribano Subías P, Barberà JA, Suberviola V. Evaluación diagnóstica y pronóstica actual de la hipertensión pulmonar. Rev Esp Cardiol 2010; 63:583-596.
206. National Pulmonary Hypertension Centers of the UK: Consensus statement on the management of pulmonary hypertension in clinical practice in the UK and Ireland. Heart 2008; 94: Suppl 1 i1-i41.

| GalerĂa de imágenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|