

Autor : Petti Marcos, Malamud Patricia, Morandi Valeria, GutiĂ©rrez Gabriela, Rivas Juan, Angiono Lucas, Nasep AgustĂn, Romero Vanesa
HIGA Eva PerĂłn, San MartĂn, Buenos Aires, Argentina Hospital Dr. Antonio Cetrángolo, Buenos Aires, Argentina
Correspondencia : Marcos Alberto Petti email: mapetti@intramed.net
Resumen
Introducción: La hipertensión pulmonar (HP) es una complicación grave que se puede
presentar en las enfermedades del tejido conectivo (ETC), múltiples causas pueden ser
el origen de la misma. Por lo tanto, es imprescindible establecer un diagnóstico preciso
para determinar la causa de la HP. Los algoritmos de diagnóstico precoz están dirigidos
a la detección temprana de la HP en pacientes con ETC
Objetivos: 1) detectar la presencia de HP en una población con diagnóstico de ETC
mediante un algoritmo de detección precoz y 2) diagnosticar HAP asociada a ETC.
Materiales y Métodos: Estudio descriptivo, prospectivo. Se incluyeron pacientes adultos
en control y seguimiento reumatológico, con diagnóstico de ETC sintomática o esclerosis
sistémica/enfermedad mixta del tejido conectivo (EMTC) sintomática como asintomática.
A aquellos pacientes con alta sospecha clínico-ecocardiográfica de HP se les realizó cateterismo cardíaco derecho (CCD).
Resultados: Se incluyeron 90 pacientes, 82 mujeres (91%) y 8 hombres (9%), con
una edad media de 52 años. Presentaban esclerosis sistémica 54 (60%), artritis reumatoidea 18 (20%), EMTC 8 (9%), lupus eritematoso sistémico 8 (9%), Sjögren 1 (1%),
polimiositis 1 (1%). La presencia de disnea fue referida en 60 pacientes (67%); un total
de 12 pacientes (13%) presentaron alta presunción de HP y se les realizó un CCD, confirmándose el diagnóstico de HP en 9 (10%), siendo 7 del grupo I HAP y 2 del grupo
II. Los pacientes con HP del grupo I se encontraban distribuidos según Clase funcional
(CF), de la siguiente manera: uno en CF III, cinco en CF II y uno en CF I.
Conclusiones: EI algoritmo utilizado permite diagnosticar HAP en pacientes con ETC
en etapas termpranas.
Palabras clave: Hipertensión pulmonar; Hipertensión arterial pulmonar; Enfermedades del tejido conectivo; Tamizaje
Application of an Algorithm for the Detection of Pulmonary Arterial Hyertension in Patients with Connective Tissue Disease
Abstract
Introduction: Pulmonary hypertension (PH) is a serious complication in connective
tissue disease (CTD), although multiple causes can be the source of it. Therefore, it is
essential to establish an accurate diagnosis to determine the cause of HP. Early detection
algorithms are aimed to diagnose PH in patients with CTD in less advanced disease.
Objectives: 1) To detect the presence of pulmonary hypertension (PH) in a population
diagnosed with CTD by an early detection algorithm. 2) To diagnose Pulmonary arterial
hypertension (PAH) associated to CTD.
Materials and Methods: It’s a descriptive, prospective study. Adult patients diagnosed with symptomatic CTD or symptomatic and asymptomatic systemic sclerosis, mixed connective tissue disease (MCTD) in the setting of rheumatologic control were
included. Those patients with high clinical and echocardiographic suspicion of PH were
catheterized (CCD).
Results: 90 patients, 82 women (91%) were included, with a median age of 52 years.
Fifty-flour (60%) corresponding to Systemic sclerosis, 18 (20%) to rheumatoid arthritis,
8 (9%) to MCTD, 8 (9%) to systemic lupus erythematosus, 1 (1%) to Sjögren and 1(1%)
to polymyositis. Dyspnea was referred in 50 patients (67%); 12 patients (13%) had high
presumption of HP and underwent a CCD, confirming the diagnosis of HP in 9 (10%).
Sever belonged to PH Group I and 2 to group II. PH group I patients were distributed
according to functional class (FC), as follows: 1 FC III, 5 FC II and 1 FC I.
Conclusions: The used algorithm allows early PAH diagnosis in CTD.
Key words: Pulmonary Hypertension, pulmonary arterial hypertension, connective tissue disease, early detection
Introducción
La hipertensión arterial pulmonar (HAP) es una
enfermedad progresiva de etiología multifactorial
que conduce al remodelado del lecho vascular precapilar y a un aumento gradual de la resistencia
vascular pulmonar (RVP) con fallo ventricular
derecho. Es una complicación grave, aunque poco
frecuente de las enfermedades del tejido conectivo
(ETC).
Se asocia con una morbilidad y mortalidad elevada a pesar de la utilización de terapias específicas
para su tratamiento1, 2.
La HAP es una forma de hipertensión pulmonar pre capilar. El diagnóstico de HAP se realiza
mediante cateterismo cardíaco derecho (CCD) y se
define como una presión arterial pulmonar media
(PAPm) igual o mayor de 25 mmHg en reposo, con
una presión de enclavamiento pulmonar wedge
(PCPw) ≤ 15 mmHg y RVP > a 3 unidades Wood
(UW), luego de excluir otras causas de hipertensión
pulmonar (HP) precapilar3.
La HAP está vinculada a un grupo heterogéneo
de entidades clínicas que comparten cambios biopatológicos similares y que han sido subcategorizadas en: idiopática, hereditaria, inducida por drogas
y toxinas, hipertensión pulmonar asociada a otras
patologías (enfermedades del tejido conectivo, hipertensión portal, infección por VIH, cardiopatía
congénita y esquistosomiasis) y recientemente
incluida dentro de este grupo la enfermedad venooclusiva y/o hemangiomatosis capilar pulmonar y
la hipertensión pulmonar persistente del recién
nacido2.
Los mecanismos exactos involucrados en la
patogenia de la HAP asociada a ETC no están
completamente esclarecidos, pero entre los mismos
se postulan: la proliferación de células endoteliales
y de células adventicias en las arterias pulmonares con crecimiento de músculo liso vascular
acompañado de la presencia de autoanticuerpos
circulantes y citoquinas pro-inflamatorias.
La esclerosis sistémica (ES) es la ETC con
mayor prevalencia de HAP (10-12% determinada
por CCD)4.
Otras comorbilidades pueden ser también responsables del desarrollo de hipertensión pulmonar
en ES, la enfermedad pulmonar difusa, la patología cardiovascular izquierda5 y la enfermedad
tromboembólica crónica entre otras. La HAP y la
enfermedad pulmonar difusa son las principales
causas de muerte en ES6, 7, 8.
Con menor frecuencia la HAP puede presentarse en pacientes con enfermedad mixta del tejido
conectivo9, lupus eritematoso sistémico10, 11 y aún
con menor prevalencia en la artritis reumatoide¹²,¹³, la dermatomiositis-polimiositis y el síndrome
de Sjögren14.
Aunque exista afectación hemodinámica temprana, los síntomas de la enfermedad vascular
pulmonar son tardíos e inespecíficos, por lo que el
diagnóstico se suele retrasar varios años15.
Los programas de detección precoz en ES para
diagnóstico de HAP han permitido diagnosticarla
en etapas más tempranas hallándose el paciente
en mejor clase funcional16, 17.
En la última década han surgido terapias específicas para el tratamiento de la HAP que mejoran su morbimortalidad18. Por lo tanto, realizar un
diagnóstico e inicio terapéutico temprano impacta
favorablemente en la evolución de la enfermedad
vascular mejorando su morbimortalidad.
Los objetivos del presente estudio son
– Detección de HP en una población con ETC.
– Diagnosticar HAP asociada a ETC.
Material y métodos
Población
Se ingresaron en forma prospectiva, y consecutiva
entre marzo del 2013 y marzo del 2014, todos los
pacientes adultos de ambos sexos, con diagnóstico de esclerosis sistémica o enfermedad mixta
del tejido conectivo sintomática o asintomática,
así como otras enfermedades del tejido conectivo
asociadas a síntomas. El diagnóstico de ETC fue
realizado por un médico reumatólogo experimentado utilizando los criterios del Colegio Americano
de Reumatología.
Para su inclusión, los pacientes debían hallarse
estables en los últimos 3 meses, con tratamiento
para la enfermedad del tejido conectivo y en ausencia de un diagnóstico previo de HP realizado
por CCD.
Se excluyeron previo al estudio a los pacientes
con enfermedad respiratoria que presentaran una
capacidad vital forzada (CVF) y/o volumen espiratorio forzado en el primer segundo (VEF1) < al 60%
del predicho y/o tomografía computada de alta resolución de tórax con anormalidad del parénquima
pulmonar o de la vía aérea relevante, pacientes con
compromiso del ventrículo izquierdo con fracción
de eyección < 45% o valvulopatía primaria moderada a severa, cardiopatías congénitas, historia de
enfermedad tromboembólica, exposición a drogas o
tóxicos y pacientes embarazadas. La espirometría
y el ecocardiograma se realizaron al ingreso del estudio y la tomografía computada de alta resolución
de tórax dentro de los 6 meses previos.
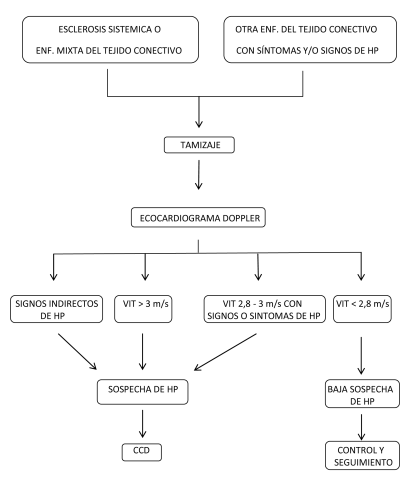
Algoritmo de detección propuesto
Se realizó un ecocardiograma doppler color a todos
los pacientes con un ecógrafo Esaote MyLab 50. Se
efectuaron todas las mediciones recomendadas por
las guías internacionales de ecocardiografía, con
el paciente en reposo en los 20 minutos previos,
prestando especial atención al estudio de las cavidades derechas19, 20, 21.
Se evaluaron síntomas y signos relacionados a
HP (disnea, síncope, presíncope, insuficiencia cardíaca derecha). La disnea se estimó según la clase
funcional (CF) de la clasificación de la NYHA. Se
registraron también los factores de riesgo, síntomas y signos relacionados a la enfermedad de base,
su tiempo de evolución y las comorbilidades. El
diagnóstico de HP se confirmó o descartó por medio
del CCD que se realizó a aquellos pacientes cuyo
ecocardiograma: doppler mostró: 1) insuficiencia
tricuspídea con velocidad pico (VIT) mayor a 3
m/s o 2) VIT entre 2,8 a 3 m/s asociado a signos
y/o síntomas de HP o 3) signos indirectos de HP
en el ecocardiograma dilatación de cavidades derechas o tronco de arteria pulmonar, hipertrofia
de ventrículo derecho (VD), tiempo de aceleración
pulmonar menor a 100 mseg, disfunción sistólica
del VD, aplanamiento del septum interventricular,
colapso sistólico del flujo de arteria pulmonar,
gradiente telediastólico de insuficiencia pulmonar
aumentado (Figura 1).
Para el diagnóstico hemodinámico de HAP, la
RVP debió ser > 3 UW. El CCD se realizó con un
catéter de Swan Ganz, con medición de presiones
intracavitarias, corrida oximétrica, volumen minuto (estimado por el método de termodilución),
resistencias vasculares e índices derivados. Dichas
mediciones se realizaron en reposo y se repitieron
en 3 oportunidades, de presentar variaciones mayores al 10% se repetían nuevamente. La correcta
posición del catéter de Swan Ganz se confirmó con fluoroscopía y con el registro de las ondas de
presión características. Todas las mediciones de
presión se realizaron al final de la espiración y en
forma manual correlacionando las ondas de presión
con el electrocardiograma.
Se definió HP con una PAPm ≥ 25 mmHg; la
misma se dividió en HP precapilar cuando presentaba una PCPw ≤ 15 mmHg y en HP postcapilar
con una PCPw > 15 mmHg.
Frente a la imposibilidad de medir en forma adecuada la PCPw se realizó un cateterismo izquierdo
con medición de la presión de fin de diástole del
ventrículo izquierdo.
En el caso de HP precapilar se realizaron los
estudios pertinentes para caracterizar el grupo
clínico y realizar el diagnóstico etiológico: estudio
funcional respiratorio completo, centellograma
pulmonar de ventilación - perfusión, serologías
para HIV y hepatitis, dosaje de hormonas tiroideas, ecografía abdominal con doppler portal, ecocardiograma con burbujas y otros estudios
considerados necesarios para descartar o confirmar
el diagnóstico.
Análisis estadístico
Para el análisis estadístico las variables continuas
se expresaron como media ± desvío estándar o
mediana con rango intercuartílico y las variables
categóricas como proporciones y/o porcentajes. En
la determinación de la distribución normal de la
muestra se empleó el test de Shapiro-Wilks. Todo
el análisis estadístico se realizó con los programas
Statistix 8.0 Analytical Software y el SPSS 10.0 Statistical análisis software for Windows (SPSS Inc.).
Resultados
Entre marzo de 2013 y junio de 2014 se evaluaron 98 pacientes con diagnóstico de ETC, se excluyeron 4 pacientes por presentar enfermedad respiratoria (2 pacientes con EPOC y 2 pacientes con fibrosis pulmonar de significación) y 4 pacientes por presentar cardiopatía con compromiso de ventrículo izquierdo (fracción de eyección < 45%). Los 90 pacientes restantes se sometieron al algoritmo diagnóstico. Las características basales de dichos pacientes se muestran en la Tabla 1.

Cuando se analizó la ES, que fue la forma más
prevalente en nuestro estudio, la misma se distribuyó de la siguiente manera: ES limitada 38/54 pacientes
(70%), ES difusa 16/54 pacientes (30%). El fenómeno
de Raynaud se presentó en 49/54 pacientes (91%). El
tiempo desde el inicio de la ETC no Raynaud hasta
el momento de la evaluación fue de 48 meses (RIQ
24-117), el tiempo de evolución del fenómeno de
Raynaud fue de 72 meses (RIQ 42-180).
En 12/90 pacientes (13%) se realizó CCD por la
alta presunción clínica y ecocardiográfica de HP.
Dicha presunción se sustentó en los siguientes
hallazgos: 8 pacientes presentaron una VIT mayor a 3 m/s, 2 pacientes con una VIT entre 2,8 y 3
m/s asociado a síntomas de HP y 2 pacientes por
presentar signos indirectos de HP. La distribución
de los mismos fue la siguiente: 9 ES, 1 artritis reumatoidea, 2 enfermedad mixta del tejido conectivo.
El diagnóstico de HP se confirmó en 9 pacientes,
de los cuales 2 presentaron HP postcapilar y 7 HP
precapilar. De este último grupo, luego de estudios
pertinentes, se confirmó que los 7 pertenecían al
grupo 1 HAP. La etiología de todos estos pacientes
con HAP fue la ES, determinando una prevalencia
de HAP asociada a ES del 13% (7/54).
La distribución según la CF de los pacientes con
HAP fue: 1 en CF III, 5 en CF II y 1 en CF I (Figura 2).
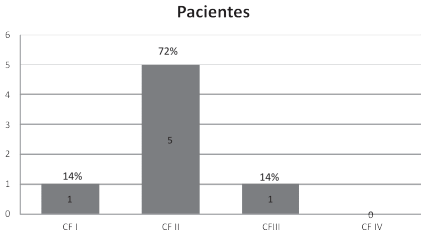
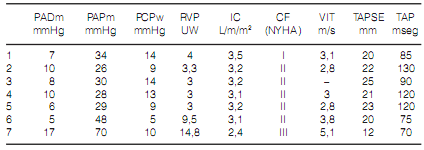
Al agrupar a los 7 pacientes con HAP en CF I/II y CF III/IV, se distribuyeron respectivamente en 6/7 (85%) y 1/7 (15%). Estos pacientes con HAP presentaron las siguientes características hemodinámicas: índice cardíaco 3,1 L/min/m2 (RIQ 2,9-3,3), RVP 3,3 UW (RIQ 3 –9,5), PAPm 30 mmHg (RIQ 28-48), presión media de aurícula derecha 8,1 ± 4,4 mmHg. De los mismos, como criterio de mayor riesgo, solo un paciente presento un índice cardíaco < 2,5 L/m/m2 y 3 presentaron una presión de aurícula derecha > 8 mmHg. Las características hemodinámicas, de CF y ecográficas más detalladas de los mismos se observan en la Tabla 2
Discusión
En el presente estudio prospectivo se observó al
aplicar un programa de detección de HP en esta
población con enfermedad del tejido conectivo, que
los pacientes se encontraban con características
clínicas y hemodinámicas favorables de su enfermedad. Su posterior caracterización y clasificación
según grupo clínico posibilita el diagnóstico de
hipertensión arterial pulmonar. En este trabajo
la mayoría de los pacientes diagnosticados se encontraban en CF I/II, con características hemodinámicas favorables (índice cardíaco > 2,5 L/m/m2,
presión de aurícula derecha < 8 mmHg) y buena
función sistólica de ventrículo derecho. El diagnóstico precoz (expresado por CF, hemodinamia y
función de ventrículo derecho) de esta patología es
de gran importancia, ya que detectar pacientes en
estadios menos avanzados de la enfermedad tiene
impacto en su evolución, ya sea por una mejor calidad de vida y/o de sobrevida con el tratamiento
disponible22.
Todos los pacientes diagnosticados con HAP
presentaban ES, esto se explica en parte porque la
ES representaba el 60% de nuestra población y la
misma es por lejos la ETC con mayor prevalencia
de HAP. Dicha prevalencia en nuestro estudio fue
del 13%, muy similar en la descripta en la literatura cuando la misma se determinó por CCD.
La utilización de un algoritmo predefinido, en
el que se incluyó la búsqueda de síntomas y signos
de HP, parámetros directos e indirectos de HP
en ecocardiografía Doppler y finalmente, el CCD
proporciona una mejor organización del proceso
diagnóstico.
El ecocardiograma doppler es el estudio complementario más utilizado para el tamizaje de HP,
tornándolo una herramienta de gran importancia
para el proceso diagnóstico, aunque no menos importante es la experiencia del ecocardiografista
para la identificación tanto de los signos directos
como indirectos de HP. En particular, el gradiente
de insuficiencia tricuspídea puede ser subestimado
si el flujo regurgitante no se encuentra bien alineado o la intensidad de la señal es débil23, 24.
El incorporar en la metodología de estudio los
síntomas y signos de HP permite utilizar distintos
valores de la velocidad de insuficiencia tricuspídea,
según estos estén presentes o no.
Los pacientes en los que se detecte síntomas y
signos de HP tienen mayor probabilidad de presentar HP y por ello en dichos pacientes utilizamos
una VIT menor.
Los valores de la VIT utilizados o recomendados
en los distintos trabajos o guías son variables, ya
que no se ha encontrado un consenso, oscilando
entre 2,5-2,8 m/s y 2,9-3,4 m/s según se asocien o
no a signos o síntomas. En algunos estudios incluso
no se utiliza la VIT y se emplean solo el valor de la
presión sistólica de arteria pulmonar (PSAP) o la
presión sistólica de ventrículo derecho (PSVD)25, 26.
En el presente estudio se utilizó el gradiente de
insuficiencia tricuspídea en lugar de la PSAP para
independizar la medición de la presión de aurícula
derecha y eliminar una de las posibles causas de
sobre o subestimación. Se decidió utilizar una VIT
mayor a 3 m/s sin síntomas o VIT entre 2,8 a 3 m/s
asociado a signos y/o síntomas de HP, en base a lo
descripto por Hachulla et al27.
El CCD continúa siendo el patrón oro para el
diagnóstico de HP y es necesario para excluir una
PCPw elevada, ya que, aunque el ecocardiograma
no evidencie disfunción diastólica de significación,
el CCD puede detectar casos de HP postcapilar
aun en pacientes con baja probabilidad de HP
postcapilar por ecocardiografía28.
Dentro de las limitaciones del estudio, se encuentran la falta de utilización de otras herramientas diagnosticas durante la evaluación inicial, como
la medición de BNP29-31 o de la capacidad de difusión
de monóxido de carbono (DLCO)32, esta última utilizada en forma aislada o como una relación con la
FVC (FVC/DLCO) utilizada en ES para evaluar el
predominio de una forma vascular o respiratoria.
La utilización del ecocardiograma como único
examen complementario para la detección de HP
también tiene sus limitaciones, como: la imposibilidad de detectar señal de IT en algunos pacientes,
lo cual impide avanzar en el diagnóstico en quienes
en general pudieran presentan HP no severa. En
los cuales es menos frecuente encontrar signos
indirectos de HP.
Otra limitación es el número de pacientes incluidos en el estudio, que fue menor que en otros
estudios internacionales multicéntricos, aunque
los resultados y conclusiones no presentan diferencias sustantivas con lo observado.
Conclusiones
El estudio realizado sugiere que, en la población analizada con enfermedad del tejido conectivo, el algoritmo diagnóstico utilizado permite detectar pacientes con hipertensión arterial pulmonar sin diagnóstico previo y en etapas más precoces de la enfermedad.
Agradecimientos: al Dr. Rodolfo Viotti por el aporte científico.
Conflicto de interés: Los autores del trabajo declaran no tener conflictos de intereses relacionados con esta publicación.
1. Condliffe R, Kiely DG, Peacock AJ, et al. Connective Tissue Disease-associated Pulmonary Arterial Hypertension in the Modern Treatment Era. Am J Respir Crit Care Med 2009; 179: 151–157.
2. Galiè N, Humbert M, Vachieryc JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2015; 46: 903-975.
3. Hoeper MM, Bogaard HJ, et al. Definitions and Diagnosis of Pulmonary Hypertension. JACC 2013; 62 (25): D 42-50.
4. Chaisson NF, Hassoun PM. Systemic Sclerosis-Associated Pulmonary Arterial Hypertension. CHEST 2013; 144: 1346-1356.
5. Fox BD, Shimony A, Langleben D, et al. High prevalence of occult left heart disease in scleroderma-pulmonary hypertension. Eur Respir J. 2013; 42: 1083-1091.
6. Nihtyanova S, Schreiber B, Ong V, et al. Prediction of Pulmonary Complications and Long-Term Survival in Systemic Sclerosis. Arthritis Rheumatol. 2014; 66: 1625-1635.
7. Hoffmann-Vold AM, Molberg O, et al. Survival and Causes of Death in an Unselected and Complete Cohort of Norwegian Patients with Systemic Sclerosis. J Rheumatol. 2013; 40: 1127-1133.
8. Mathai SC, Hummers LK, Champion HC, et al. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: impact of interstitial lung disease. Arthritis Rheum. 2009; 60: 569-577.
9. Gunnarsson R, Andreassen AK, Molberg O, et al. Prevalence of pulmonary hypertension in an unselected, mixed connective tissue disease cohort: results of a nationwide, Norwegian cross-sectional multicentre study and review of current literature. Rheumatology 2013; 52: 1208-1213.
10. Prabu A, Patel K, Yee CS, et al. Prevalence and risk factors for pulmonary arterial hypertension in patients with lupus. Rheumatology 2009; 48: 1506-1511.
11. Ruiz-Irastorza Guillermo, Garmendia Maider, Villar Irama, et al. Pulmonary hypertension in systemic lupus erythematosus: prevalence, predictors and diagnostic strategy. Autoimmun Rev. 2013; 12: 410-415.
12. Dawson J.K, Goodson N.G, Graham D.R, Lynch M.P. Raised pulmonary artery pressures measured with Doppler echocardiography in rheumatoid arthritis patients. Rheumatology 2000; 39: 1320-1325.
13. Keser G, Capar I, Aksu K, et al. Pulmonary hypertension in rheumatoid arthritis. Scand J Rheumatol. 2004; 33: 244-245.
14. Launay D, Hachulla E, Hatron PY, Jais X, Simonneau G, Humbert M. Pulmonary arterial hypertension: a rare complication of primary Sjögren syndrome: report of 9 new cases and review of the literature. Medicine (Baltimore). 2007; 86: 299-315.
15. Escribano-Subias P, Blanco I, Loóez-Meseguer M, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J 2012; 40: 596-603.
16. Humbert M, Yaici A, De Groote P, et al. Screening for Pulmonary Arterial Hypertension in Patients with Systemic Sclerosis Clinical Characteristics at Diagnosis and Long-Term Survival. Arthritis&Rhematism 2011; 63: 3522-3530.
17. Khanna D, Gladue H, Channick R, et al. Recommendations for Screening and Detection of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Arthritis Rheum. 2013; 65: 3194-3201.
18. Launay D, Sitbon O, Le Pavec J, et al. Long-term outcome of systemic sclerosis-associated pulmonary arterial hypertension treated with bosentan as first-line monotherapy followed or not by the addition of prostanoids or sildenafil. Rheumatology (Oxford). 2010; 49: 490-500.
19. D’Andrea A, Naeije R, Grünig E, et al. Echocardiography of the Pulmonary Circulation and Right Ventricular Function Exploring the Physiologic Spectrum in 1,480 Normal Subjects. CHEST 2014; 145: 1071-1078.
20. Karasa MG, Kizera JR. Echocardiographic assessment of the right ventricle and associated hemodynamics. Prog-Cardiovasc Dis. 2012; 55: 144-160.
21. Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the Echocardiographic Assessment of the Right Heart in Adults: A Report from the American Society of Echocardiography Endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010; 23: 685-713.
22. Mukerjee D, St George D, Coleiro B, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003; 62: 1088-93.
23. Borgeson D, Seward J, Miller FJ, et al. Frequency of Doppler measurable pulmonary artery pressures. J Am SocEchocardiogr.1996; 9:832-837.
24. Guerra Ramos FJ. Papel de la ecocardiografía ante la sospecha de hipertensión pulmonar. Arch Bronconeumol. 2011; 47: 7-11.
25. Galiè N, Hoeper MM, Humbert M, et al. Guía de práctica clínica para el diagnóstico y tratamiento de la hipertensión pulmonar. Rev Esp Cardiol. 2009; 62: 1464. e1-e58.
26. Phung S, Strange G, Chung LP, et al. Prevalence of pulmonary arterial hypertension in an Australian scleroderma population: screening allows for earlier diagnosis. Intern Med J. 2009 2009; 39: 682-691.
27. Hachulla E, Gressin V, Guillevin L, et al. Early Detection of Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis & Rhematism. 2005; 52: 3792-3800.
28. Saggar R, Sitbon O. Hemodynamics in Pulmonary Arterial Hypertension: Current and Future Perspectives. Am J Cardiol. 2012; 110: 9S-15S.
29. Williams MH, Handler CE, Akram R, etal. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arteria lhypertension. Eur. Heart J 2006; 27: 1485-1494.
30. Allanore Y, Borderie D, Avouac J, et al. High N-terminal pro-brain natriuretic peptide levels and low diffusing capacity for carbon monoxide as independent predictors of the occurrence of precapillary pulmonary arterial hypertension in patients with systemic sclerosis. Arthritis Rheum 2008; 58: 284-291.
31. Mukerjee D, Yap LB, Holmes AM, et al. Significance of plasma N-terminal pro-brain natriuretic peptide in patients with systemic sclerosis-related pulmonary arterial hypertension. Respir Med 2003; 97: 1230-1236.
32. Mukerjee D, St George D, Knight C, et al. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology. 2004; 43: 461-466.

| GalerĂa de imágenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|