

Autor : Brenda Varela, Gabriela Tabaj, Juan Ignacio Enghelmayer, Silvia Quadrelli, Tulio Papucci, Andrea Pino, Marcelo Fernández Casares
AAMR Department of Interstitial Diseases
Correspondencia : Brenda Varela Address: Pueyrredon Av 2127 - CABA. Phone: 4-8277000 (2722) E-mail: brendavarela144@hotmail.com
Abstract
Background: Diffuse interstitial (or parenchymal) lung diseases (ILDs) are a very large
group of diseases that although they share certain clinical features, have a very different
prognosis. Idiopathic pulmonary fibrosis (IPF) is the most prevalent in many countries
and its diagnosis can be difficult. After the results shown in the consensus on diagnosis
and management of IPF, and the arrival of new drugs such as pirfenidone, the approach
to this disease have changed. A survey was performed to argentine pulmonologists in
order to evaluate the acceptability and implementation of these guidelines in Argentina.
Material and Methods: A survey of 24 questions was designed. Among the data collected
in the questionnaire were demographics of respondents, workplace (public or private
healthcare facilities, referral center, large or small healthcare centers or institutions),
frequency at which IPF patients were examined, availability of diagnostic tests, and
diagnostic strategies used with ILD patients. Finally, the survey focused on therapeutic
recommendations for patients diagnosed with IPF. The survey was completed during the
Argentine Congress of Respiratory Medicine held in 2013 in the city of Mendoza. The
same methodology and questionnaire were previously used in the Argentine Congress
of Respiratory Medicine in 2011.
Results: In 2013, a total of 252 physicians completed the survey, which represented approximately 20% of Congress attendees. The complementary test of higher availability
was the the six minutes walk test (6MWT). The most widely used supplementary method
was thoracic computed tomography (CT) as 86.9% of the responders used it if they
suspected ILD, and only 44.4% of the responders used diffusing capacity of the lungs for
carbon monoxide (DLCO) with all their patients. Almost 50% of the responders consulted
referral centers for less than 30% of patients with suspected ILD. Less than 20% of the
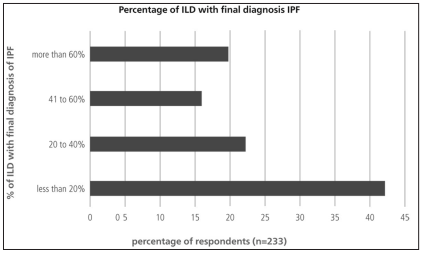
responders considered that they reached a final diagnosis of ILD in over 60% of their
patients. Final distribution of diagnosis was heterogeneous. Interestingly, almost 50% of
the responders considered IPF as the fnal diagnosis in less than 30% of their patients.
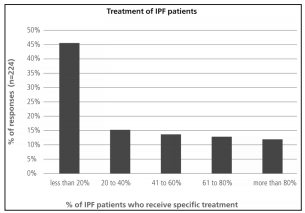
Approximately 50% of the responders answered that less than 20% of their IPF patients
received specific treatment for the disease.
Conclusions: Our survey suggests that there are difficulties in the diagnostic approach
of ILDs, there is a low percentage of patients that are evaluated in referral centers and
there is a low proportion of IPF patients receiving specifc treatment.
Key words: Diffuse interstitial (or parenchymal) lung diseases; Idiopathic pulmonary fibrosis; Connective Tissue Disease; Pirfenidone
Introduction
Diffuse interstitial (or parenchymal) lung diseases
(ILDs) represent a very large group of more than
200 entities, many of which are rare or “orphan” diseases. The ATS/ERS (American Thoracic Society/ European Respiratory Society)1 consensus
classifies idiopathic interstitial pneumonias (IIP)
into seven clinical-pathological conditions. This
classification –largely based on histopathology– is
also the result of a close relationship among clinicians, radiologists, pathologists and a multidisciplinary approach. High resolution thin-section CT
scanning (HRCT) is a key tool for IIP diagnosis
since in most cases, in the adequate clinical context, tomographic patterns are enough to reach
final diagnosis.
Idiopathic pulmonary fibrosis (IPF) is a progressive, fibrosing disease. It does not have a
cure and mean survival ranges from 2 to 5 years
from diagnosis2. Many clinical research studies
looked for an effective treatment without success
until 2011, when the first treatment for IPF was
approved in Europe: pirfenidone3.
ILD diagnosis can be very difficult, especially
when it is an IPF or a nonspecific interstitial pneumonia (NSIP). Several societies (ATS, ERS, JRS
and ALAT) have extensively reviewed available
literature and guidelines on recommendations for
IPF diagnosis and management. New information
has become available after 2011 with the approval
of pirfenidone and the negative outcomes of the “triple drug therapy”- consisting of corticosteroidsazathioprine and N-acetylcysteine4.
Acceptability and difficulties regarding the
implementation of these guidelines have not been
assessed in Argentina.
To assess the parameters used in IPF diagnosis
and treatment, a survey was conducted among
pulmonologists attending the Argentine Congress
on Respiratory Medicine held in October 2013. The
results of this survey were compared to those of a
similar survey carried out two years earlier by the
Interstitial Disease Department of the Argentine
Association of Respiratory Medicine.
Materials and methods
A 24-item questionnaire was designed to characterize pulmonologists current clinical practice
patterns regarding availability of resources, assessment of ILDs, and treatment of IPF patients.
During the Argentine Congress of Respiratory
Medicine held in October 2013 in the city of Mendoza, the participants were invited to complete the
questionnaires that were placed on a desk. The
following information was collected: demographics
data of respondents, workplace (public or private
healthcare facilities, referral center, large or small
healthcare centers or institutions), frequency at
which IPF patients were examined, availability
of diagnostic tests, and diagnostic strategies used
with ILD patients. Finally, the survey focused
on therapeutic recommendations for patients
diagnosed with IPF. The same methodology and
questionnaire had been used previously at the
2011 Argentine Congress of Respiratory Medicine.
Data were presented as proportion of responders.
The chi=square test was used for the comparison
between physicians who devote more than 50% of
their time working at high-complexity healthcare
facilities and those working at small healthcare
centers. Comparisons between 2013 and 2011 responders were also done using a chi-square test.
ANNEX I and II surveys in spanish version.
Results
In 2013, a total of 252 physicians completed the
survey, which represented approximately 20% of
Congress participants. In 2011, 155 physicians
answered the questionnaire.
Over 60% of 2013 responders were women
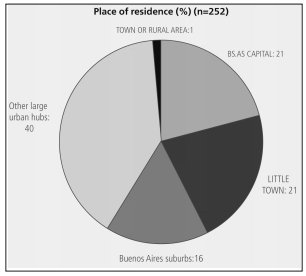
under 50 years old (Table 1) who lived in large
cities (Figure 1). 90% were pulmonologists (12%
were specialists in a related field, such as internal
medicine or intensive health care). 29% of the
responders stated that they worked (more than
50% of their time) at public centers and 25% in a
private referral centre. The remaining stated that
they worked at small or private institutions. Over
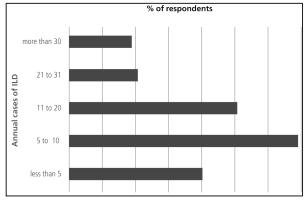
80% of responders were involved in less than 20
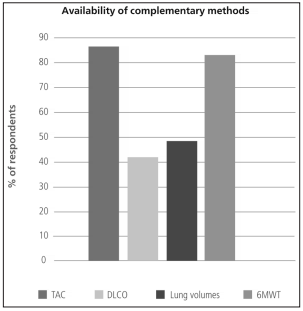
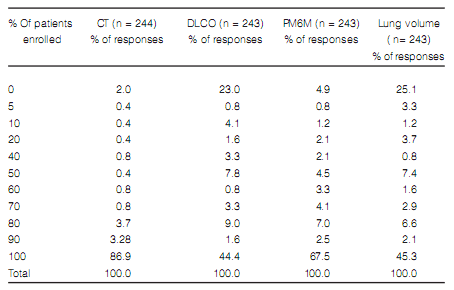
annual cases of ILD (Figure 2). The most available complementary method was the six minutes walk
test (6MWT) (Figure 3). The most widely used
supplementary method was thoracic computed tomography (86.9% of the responders used it if they
suspected ILD), and only 44.4 % of the responders
used lung diffusing capacity for carbon monoxide
(DLCO) in all their patients (Table 2). Table 3 shows the frequency of other diagnostic methods
which were used.
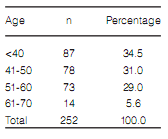
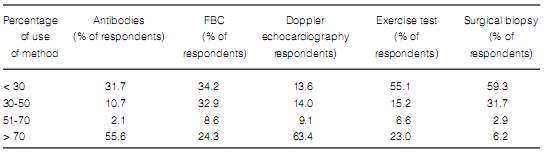
Reasons for not using some diagnostic methods varied, but a high percentage of respondents stated that the limiting factors to order some tests were high cost and unavailability (Table 4).
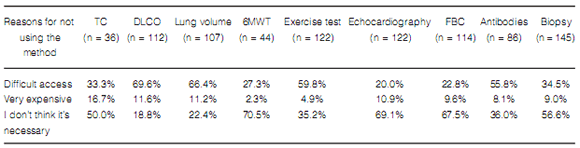
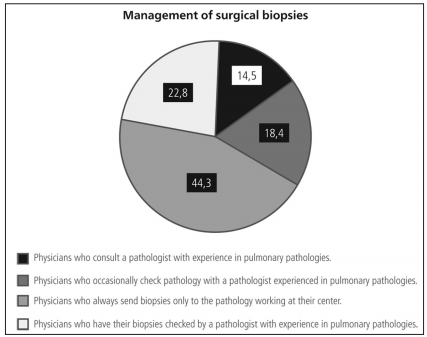
If surgical biopsies were performed, approximately one third of the responders routinely sent their samples to an expert in pulmonary pathology (Figure 4). Almost 60% of the respondents consulted referral centers for less than 30% of their patients with suspected ILD (Figure 5). Among the 190 respondents who explained their reasons for this, 58.4% stated they considered it unnecessary, and 41.5% said they had limited access to specialized centers.
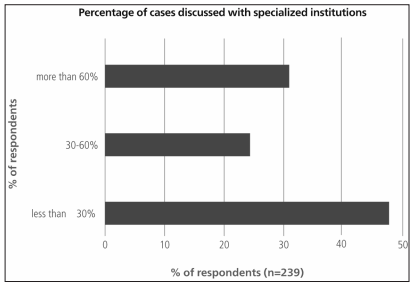
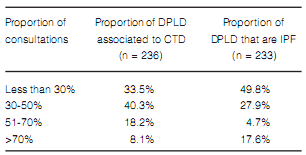
Less than 20% of the responders considered that they reached a final diagnosis of ILD in over 60% of their patients (Figure 6). Final classification of diagnosis was heterogeneous. It is worth noting that, almost 50% of the respondents considered IPF as the final diagnosis in less than 30% of their patients (Table 5). Among the alternative diagnoses it had a large proportion of interstitial disease associated with collagen diseases. Approximately 50% of the responders answered that less than 20% of their IPF patients received specific treatment for the disease (Figure 7).
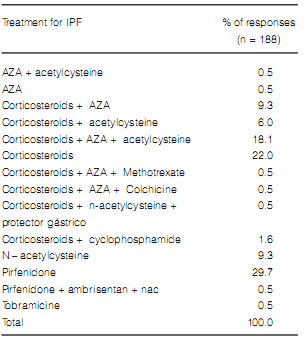
Only 30% of the responders prescribed pirfenidone as the choice treatment for IPF, and over 60% continued prescribing to prescribe treatments with different combinations of corticosteroids and immunosuppressants (Table 6). Over 90% of the responders expressed that less than 3 patients per year were referred for transplant evaluation. Reasons for not referring patients were as follows: transplant was not considered a treatment option (3.9%), there were few severe patients in their population (72.5%) and the belief that getting a lung transplant in our country is extremely unlikely (23.5%).
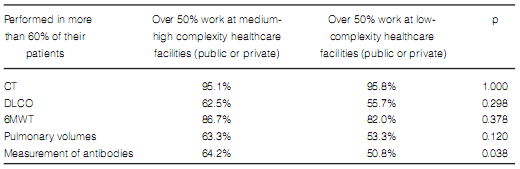
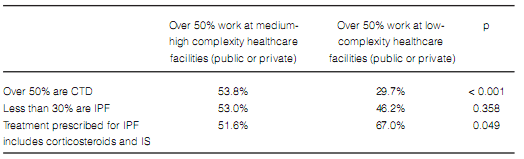
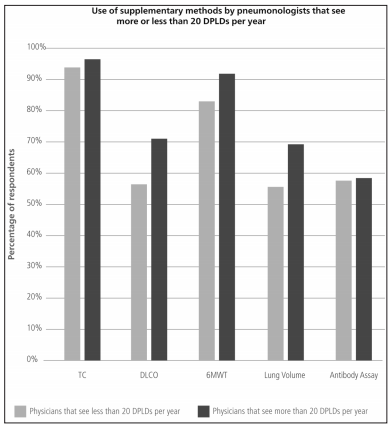
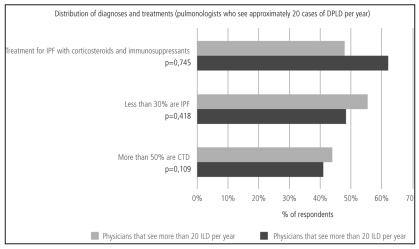
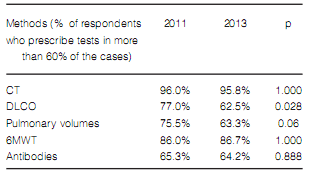
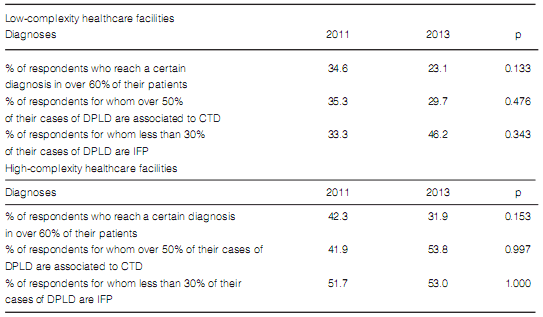
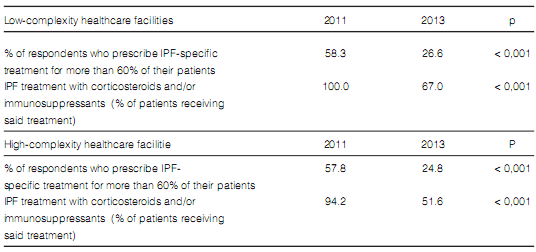
The frequency in the indication of antibody assays was the only difference found in terms of diagnostic methods when comparing pulmonologists who worked more than 50% of their time at medium and high-complexity facilities with those working at low-complexity facilities or small healthcare centers (Table 7). Pulmonologists at large healthcare facilities reported a higher prevalence of ILD associated to connective tissue disorders and a lower rate of prescription of corticosteroids and immunosuppressants for the treatment of IPF (Table 8). There were no differences between pulmonologists who took care of more than 20 ILD cases per year and those who took care of less cases (Figures 8 and 9).
The 2011 and 2013 results were not entirely
comparable, since the prevalence of the responders who worked more than 50% of their time
at referral centers was higher in 2011 (64.7% vs.
50.2% p: 0.004). Therefore, we decided to compare
according to complexity.
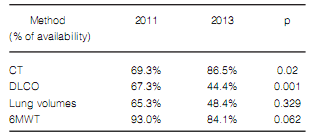
We confirmed a reduced availability of DLCO
and, therefore, in DLCO measurement between
2011 and 2013 in the surveyed centers workingin medium and high complexity (Tables 9 and
10). In both types of institutions, the classification of the final diagnoses was not different
(Table 11).
The proportion of patients receiving corticosteroids and/or immunosuppressants for the treatment of IPF decreased significantly. However, this decrease was neither accompanied with nor replaced by new treatment options (Table 12).
Discussion
During the last few years, the diagnosis of interstitial diseases –particularly IPF– has undergone
many changes. This may be partly due to a deeper
understanding of its physiopathology, the advent of
new drugs, and, mainly, the publication of the last
2011 consensus1 on IPF diagnosis and treatment.
In addition to the strengths and weaknesses of
different therapeutic options, there was a turning point in the management of these patients marked
by the contribution of HRCT to diagnosis –often
with high certainty– and the need for an interdisciplinary approach to these patients.
Although several countries have assessed the
impact of these guidelines in the management of
IPF patients3,5, 6, 7, 8, their acceptability and implementation of the mentioned guidelines have not
been assessed in our country. In order to evaluate the impact of the 2011
Consensus1 in our country, a questionnaire was
designed to be administered during the 2011 and
2013 sessions of the annual Argentine Congress
of Respiratory Medicine, an event that is attended
by many physicians.
Regarding the study population (253 surveyed
physicians in 2013 and 155 in 2011), we must
consider that slightly over half of responders worked at public or private referral centers, while
the rest worked exclusively in private practice or
small healthcare facilities. This is an important fact, since (as mentioned before) diagnosis of most
interstitial diseases requires a multidisciplinary
approach, which calls for access to and the interaction of different specialists (pulmonologists,
imaging experts, and pathologists). Such approach
allows for earlier diagnosis (as in IPF) and adequate treatment decisions9. The workplace of
surveyed physicians contrasts with international surveys. According to a recently published French
survey5 –which polled over 1244 pulmonologists,
out of whom, 41% dealt with interstitial diseases–most pulmonologists worked at general hospitals
(68%), 23% in private practice, and 9% in combined
private/public practice.
In a survey conducted in Latin America-ALAT
2013 (Latin American Thoracic Society)10, a total
of 185 healthcare professionals were polled (most
of them pulmonologists), 76% of whom were over
40 years of age. In our survey, over 60% of the
respondents were women under 50. This means
that in both surveys, physicians had at least 10
years of experience in their specialty.
In the 2013 survey, the analysis of the number
of IPF patients assisted per year showed that 80%
of respondent physicians saw less than 20 patients
with ILD per year (55% saw 10 or less per year).
This is a significant difference with international
surveys; the French5 and AIR3 (Advancing IPF
Research) surveys showed mean values of 56 and
39 IPF patients per physician per year, respectively.
In the Latin American survey10, 50% of the respondents stated that 1-5% of their patients had ILD.
Accordingly, Argentina´s geographic dispersion
and low stratification of complexity levels call for
continuous medical education strategies for ILD
that require support structures (on-site or remote)
for a large number of physicians working at centers with low availability of diagnostic methods
and limited access to interdisciplinary discussions.
As regards availability of supplementary studies, 86.9% of respondents had access to CT, 67% to 6MWTs, and only 44% to DLCO. The Latin American survey10 showed very similar results: 85% for
CT and 40% for DLCO. HRCT is very important in
the diagnosis of IPF, since a tomographic pattern of
usual interstitial pneumonia (UIP) would avoid the
need for surgical biopsy confirmation. Our study
did not enquire about access to experts in thoracic
images nor how the diagnosis was performed in
patients without CT.
In Argentina, there are very few official graduate programs on thoracic radiology. However,
this situation is not conclusive since radiologists
may have attended other continuous medical education programs or have acquired extensive
experience because of the number of cases they
have seen. Even though it is very hard to assess
this situation quantitatively, it is unlikely that
the physicians working at small centers (and/or
small districts) have direct access to a radiologist
with the opportunity to see a large number of
cases, as would be necessary to upgrade their
experience beyond their initial training. Another
limiting factor of this type of survey is that it cannot assess imaging quality (especially in the case
of high-resolution techniques) and, accordingly,
diagnostic accuracy.
As for supplementary tests, it is worth mentioning that a relatively low proportion of physicians
perform DLCO routinely in all their patients
(44%), which is probably due, as referred by responders, to lack of availability (69%) and/or high
cost (11.6%). DLCO is essential to assess disease
severity and surgical risk –in case of biopsies.
Remarkably, with respect to their reasons for not
conducting supplementary tests, they answered
that they thought they were not necessary (18.8%).
In spite of the fact that, in theory, access to 6MWT
is easy (it is a simple, low-cost test), only 67.5%
of the respondents used it routinely, and 70% of
non-users stated they considered it unnecessary.
6MWT is extremely useful both to detect baseline
defects –if DLCO is not available– and to predict disease evolution in IPF patients. Other local studies
have shown that the test is not sensitive enough to
detect a drop in DLCO11 but it is readily available
and highly-specific. On the other hand, the drop in
SaO2 correlates to the drop in forced vital capacity
(FVC) but it cannot be predicted by DLCO values
or thoracic CT anomalies. Thus, that measurement
is important to decide whether or not to administer
supplemental oxygen11. Additionally, 6MWTs have
proved useful to define IPF prognosis, whether
through distance walked or the magnitude of the
drop in oxygen saturation12. A study13 showed that
the drop in oxygen saturation (more than the distance walked) predicted a higher mortality risk,
even if adjusted to age, sex, and baseline DLCO.
Another study14 showed that walked distance was
not effective to predict a higher mortality risk,
if the drop in oxygen saturation was taken into
consideration. These differences probably relate
to different walking protocols and the use or
absence of supplemental oxygen but, in any case,
the diagnostic and prognostic value of this simple
affordable test is undeniable. Strikingly, we found
no significant differences between physicians working at medium-high complexity healthcare facilities (and/or who saw more patients per year) and
the remaining healthcare professionals regarding
the use of routine tests—with the only exception
of antibody assays, which were more frequently
prescribed by the first group of physicians.
As for interdisciplinary diagnosis, only one third
of responders sent (always or frequently) biopsy
samples to pathologists with experience in interstitial diseases (37%). This differs from other surveys,
since in AIR this is a systematic practice in over 90% of the cases (66% in the French survey). It is worth
highlighting that, in those surveys, only 7% and
3%, respectively, never consult another colleague
(of the same field or radiologists or pathologists)
for help to reach IPF diagnosis. In the Latin American survey10, more than half of the responders
(59%) did not have access to a pathologist with
experience in ILD diagnosis in their institution.
Such data, both for Argentina and Latin America,
lead us to consider it necessary to promote or facilitate communication with referral centers for ILD
management. Although different characteristics of
the sample population account for these differences,
difficulties to refer laboratory samples undoubtedly
have a large influence in our settings. To promote
and guarantee access to experienced pathologists
should be a priority for parties interested in these
patients, so as to facilitate the work of specialists
without access to pathologists trained in pulmonary
pathologies at their centers.
In our survey, less than 50% of responders
consult referral centers. Maybe this low rate of
interaction partially explains why only 20% reach
final diagnosis in their patients. In Argentina,
there are free services and methods for face-to-face
and remote consultation with specialized centers.
However, consultation rates to specialized centers are very low, apparently due to lack of trust
or knowledge. The Department of Interstitial
Diseases of the AAMR (Argentine Association for
Respiratory Medicine) should improve accessibility
to and promote the importance of these consultations more broadly and emphatically—especially
in difficult-to-diagnose cases.
Another striking aspect is the low proportion
of physicians stating that IPF is their most frequent diagnosis. Instead they claimed a higher
frequency of connective tissue diseases (CTDs).
The assessment of different international series
(France, Japan, USA, etc.) shows that IPF is the
most prevalent diagnosis, representing over 50%
of ILD cases. This was also the result of the Latin
American survey, where the most frequent ILD
was IPF (59%). This situation may result from
the inclusion, within the ILD category, of patients
with non-specific disorders or conditions not traditionally included within ILD, such as infections,
occupational conditions, or a high level of wrong
diagnoses. However, except for specialized hospitals, the low prevalence of ILDs associated to CTDs
makes it unlikely for CTDs to be the most frequent cause. A positive aspect of this information is the
high awareness among rheumatologists who refer
their patients frequently and early for evaluation
of lung involvement, though, this may also reflect
the under-diagnosis of IPF.
It is worth mentioning the low percentage of
patients who receive specific treatment. Less than
one third of physicians prescribed pirfenidone as
the choice treatment. Pirfenidone is currently
regarded as the only drug available in the Argentine market with positive, proven results. The
ASCEND (Assessment of Pirfenidone to Confirm
Efficacy and Safety in Idiopathic Pulmonary
Fibrosis) 15 study is a phase-III, double-blind,
placebo controlled randomized trial assessing
the use of pirfenidone in the IPF treatment. In
a study, conducted for the first time in Japan,
treatment with pirfenidone reduced the decline
in FVC at week 52 and improved survival. Later,
multicenter studies were conducted in the US
(CAPACITY 004 and 006)16, where the primary
outcome was FVC (difference between baseline
and end values at week 72). The difference was
reached in 004 but not in 006. Treatment with
pirfenidone was associated to a reduction in FVC
decline (8 vs. 12%) as well as to fewer deaths in
patients treated with 2403 mg/d. This is why several international guidelines have included pirfenidone in their treatment recommendations17,18,19.
A more concerning issue is the fact that 60% of
the responders, continued to prescribe different
combinations of corticosteroids and immunosuppressants, which not only are ineffective but may
be harmful. The study known as PANTHER-IPF20 assessed the response of IPF to three different
therapeutic regimens: prednisone, azathioprine
and n-acetylcysteine versus n-acetylcysteine
alone versus placebo. An interim analysis conducted before week 60 showed that, as compared
to placebo, the combination of the three drugs
(prednisone, azathioprine and n-acetylcysteine)
was associated to higher mortality rates (1% vs.
11%), more hospital admissions (8% vs. 29%), and
more severe adverse events (9% vs. 31%).
In the Latin American survey, most colleagues
still used the triple drug regimen as a therapeutic
option. In the AIR survey (conducted in 2013),
pirfenidone was prescribed to 81% of IPF patients,
being the most frequent prescription after supplemental oxygen (96%). N-acetylcysteine alone and
corticosteroids were prescribed to 76% and 34%, respectively. It seems that high-cost and availability difficulties have a significant influence in
our settings, limiting treatment administration.
However, it is also possible that the fact that IPF
is perceived as an inevitably lethal disease in the
short-term influences treatment decisions made
by treating physicians, explaining why they do not
take a more active approach with these patients. It
is necessary to effectively communicate the correct
role of pirfenidone (with its indications, contraindications, and effectiveness limitations) so that
both physicians and patients can make informed
decisions about the use of pirfenidone and future
available treatments to slow disease progression.
According to the French survey (2011-2012),
treatment with corticosteroids was prescribed in
49% of the cases. This high proportion—observed both in the French survey and in our own
survey—leads us to emphasize the need for continuous education and refreshing programs on IPF
management. Another factor worth mentioning
(which we should not forget) is that pirfenidone
has been available in our country only since 2013-
a relatively short time. Factors such as resistance
to change, lack of knowledge about the consensus
and new drugs, as well as delays of public and
private health insurance companies in covering
costs, could account for this.
In the comparison between 2011 and 2013
surveys, the reduction in the availability and,
therefore, the use of DLCO for ILD patients called our attention. This poses a paradox, since
it is well known that during the last few years
the availability of high-complexity pulmonary
function tests has increased in Argentine provinces. Another relevant piece of information that
derives from such comparison is the reduction
in the use of corticosteroids and immunosuppressants (probably due to the dissemination
of information about their toxicity), but this
was not accompanied by a higher use of specific
treatments, since the prescription rate of specific
treatments was also low. However, it is highly
worrying that even Argentine pulmonologists
(51.6% of whom work at large hospitals and
high-complexity facilities vs. 67% who work at
low-complexity healthcare facilities) who completed the survey (half of whom stated they saw
quite a few cases of IPF patients per year) still
prescribe corticosteroids and immunosuppressants, a treatment proven to be ineffective and highly unadvisable because it is associated with
increase in mortality rates20.
The publication of the 2011 Consensus1 and
continuous medical education programs conducted by the Argentine Association of Respiratory
Medicine have not yet had the expected impact
in reducing the prescription of useless drugs or
encouraging interdisciplinary consultations. Behavioral changes as regards diagnosis and treatment
(significantly different in the literature and the
community of specialists) associated to the publication of the consensus and pivotal studies on
the different treatments used have not changed
the therapeutic decisions of Argentine surveyed
physicians as expected. Obviously, emphasizing
the need for continuous education programs in
the management of these diseases is of paramount
importance for any program aimed at improving
care for these patients.
Among the limitations of this survey, we have to
state, among others, low percentage of answers for
some items and the responders arbitrary criteria to
define high-complexity and low-complexity medical
centers. Comparisons with other surveys are based
only on estimates, since designs and populations are
different. In addition to this, as mentioned before,
the surveys included physicians engaged in the
management of interstitial diseases.
However, it is worth pointing out that the number of responders represents almost one third of
Argentine pulmonologists, since it is estimated
that there are approximately between 900 and
1000 pulmonologists in our country.
The new challenge is to assess and identify the
underlying causes of the barriers to diagnosis
and treatment of these diseases in our setting.
Promoting benchmark centers, creating surveys
to identify problems, translating literature into
our language, and using technology for on-line
communication (e-mail or videoconference) could
mark the start of a better implementation of recent
international practice guidelines.
Acknowledgements: The authors want to acknowledge the collaboration of Dr. María Otaola, Dr. Valentina Di Boscio, Dr. Andrea Werbach, Dr. Liliana Castro Zorrilla, Dr. Santiago Rossi, Dr. Adrian Gaser, Dr. Fabian Caro, Dr. Gloria Olmedo, Dr. Luciana Molinari, Dr. Mirta Scarinci, Dr. Glenda Ernest, Dr. Flavia Logrado, Dr. Carlos Mosca, Dr. Raúl Darío Rey, Dr. Mariano Mazzei, Dr. J. Antonio Mazzei, Dr. Juan C. Spina, Dr. Nora Falcoff, Dr. Haydeé Gutiérrez, Dr. Lilian Capone, Dr. Liliana Dalurzo, who “on behalf of AAMR’s Department of Interstitial Diseases” contributed to preparing and analyzing this survey. The authors also want to thank AAMR 2012-2013 Board of Directors for their unlimited support to this project.
Conflict of interest: The study was partially sponsored by DOSA laboratories. GT has received funding from GSK, BI, DOSA to attend conferences; is part of a protocol in IPF and pirfenidone funded by DOSA; speaker of BI and Bristol Myers Squibb. AP is employed by Novartis Argentina. BV has received funding for conferences and internships for Novartis, Raffo and DOSA. MFC is part of a protocol in IPF and pirfenidone funded by DOSA. JIE has received funding to attend conferences by Novartis, Montpellier, Phoenix and Roemmers; is a scientific advisor of Novartis. TP has received funding to attend conferences by DOSA, Boehringer, Bayer, Glaxo, Temis Lostalo and Biotoscana. SQ is a consultant in the research study “Safety of pirfenidone in IPF” sponsored by DOSA and speaker of BI.
1. Raghu G, Collard HR, Egan JJ et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788-824.
2. Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431-440.
3. Cottin V. Current approaches to the diagnosis and treatment of idiopathic pulmonary fibrosis in Europe: the AIR survey. Eur Respir Rev 2014; 23(132): 225-30.
4. McGrath EE, Millar AB. Hot off the breath: triple therapy for idiopathic pulmonary fibrosis-hear the PANTHER roar. Thorax 2012; 67(2): 97-8.
5. Collard H, Loyd J, King TE Jr et al. Current diagnosis and management of idiopathic pulmonary fibrosis: A survey of academic physicians. Respiratory Medicine 2007; 101: 2011-2016.
6. Peikert T, Daniels C, Beebe TJ et al. Assessment of current practice in the diagnosis and therapy of idiopathic pulmonary fibrosis. Respiratory Medicine 2088; 102: 1342-1348.
7. Bando M. A prospective survey of interstitial idiopathic pneumonias in Japan. Article in press.
8. Cottin V, Cadranel J, Crestani B et al. Management of idiopathic pulmonary fibrosis in France: A survey of 1244 pulmonologists. Respiratory Medicine 2014; 108: 195-202.
9. Do Bois R. An early and more confident diagnosis of idiopathic pulmonary fibrosis. Eur Respir Rev 2012; 21: 124, 141-146.
10. Curbelo P. Encuesta de EPID en Latinoamérica-ALAT 2013. Respirar 2013; 5:2 5-8.
11. Quadrelli S, Ciallella L, Catalán Pellet A et al. Compromiso pulmonar en esclerosis sistémica. Medicina (B. Aires) 2007; 67: 5.
12. Molinari L, Quadrelli S, Tabaj G et al. Factores predictores de la caída de la saturación de oxígeno durante la caminata de 6 minutos en la fibrosis pulmonar idiopática. Rev Am Med Resp 2009; 9: 175-180.
13. Lama VN, Flaherty KR, Toews GB et al. Prognostic value of desaturation during a 6- minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003; 168: 1084-90.
14. Flaherty KR, Andrei AC, Murray S et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174: 803-9.
15. King TE, Bradford WZ, Castro-Bernardini S et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2083-92.
16. Noble PW, Albera C, Bradford WZ et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377: 1760-1769.
17. Idiopathic pulmonary fibrosis: The diagnosis and management of suspected idiopathic pulmonary fibrosis. Nice Guidelines 2013.
18. Cottin V, Crestani B, Valeyre D et al. Diagnosis and management of idiopathic pulmonary fibrosis: French practical guidelines. Eur Respir Rev 2014; 23(132): 193-214.
19. Behr J, Günther A, Ammenweth W et al. German guideline for diagnosis and management of idiopathic pulmonary fibrosis. Pneumologie 2013; 67(2): 81-111.
20. The Idiopathic Pulmonary Fibrosis Clinical Research Network. N Engl J Med 2012; 366: 1968-1977.

| GalerĂa de imágenes | ||
| Mujer joven con afectaciĂłn pulmonar bilateral y alteraciĂłn de la conciencia | ||
Autores: Churin Lisandro |
 |
|